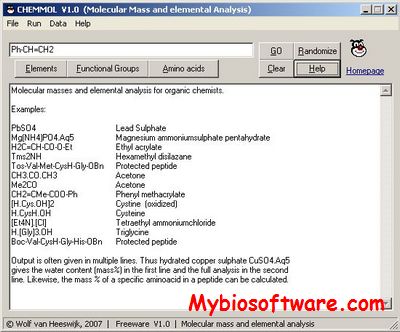
CubeX
:: DESCRIPTION
CubeX calculates haplotype frequencies using the exact solution to the cubic equation rather than an iterative approach.
::DEVELOPER
Tom Gaunt’s group in the MRC IEU
:: SCREENSHOTS
N/A
:: REQUIREMENTS
- Linux /Mac OsX / Windows
- Python
:: DOWNLOAD

:: MORE INFORMATION
Citation:
Gaunt TR, Rodríguez S, Day IN.
Cubic exact solutions for the estimation of pairwise haplotype frequencies: implications for linkage disequilibrium analyses and a web tool ‘CubeX’.
BMC Bioinformatics. 2007 Nov 2;8:428.