TAMO 20120321
:: DESCRIPTION
TAMO (Tools for Analysis of MOtifs) is developed around a unified motif representation of a position-specific scoring matrix (PSSM). Motif objects may be assembled from IUPAC-ambiguity codes, multiple sequence alignments, averages of other motifs, and matrices of frequencies or log-likelihood values. Motifs can printed, concatenated, indexed and sliced like text strings, or rendered as sequence logos. They can also be randomized, reverse-complemented, and recomputed using different assumptions about background base frequencies. Motifs can also store and report information about their origin, information content, and score. Finally, motifs can scan DNA sequences for instances of matching sites.
::DEVELOPER
The Fraenkel Lab
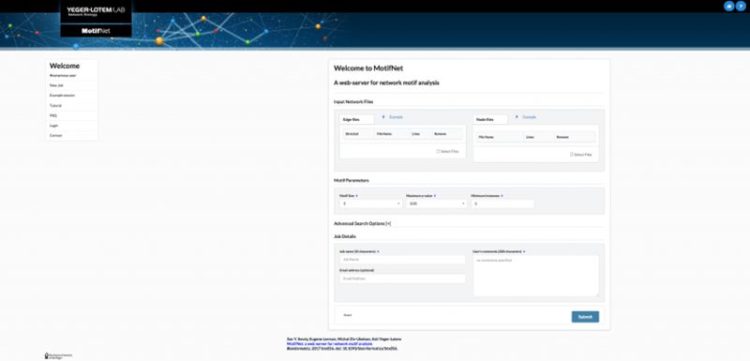
:: SCREENSHOTS
N/A
:: REQUIREMENTS
- Linux / Windows/ MacOsX
- Python
:: DOWNLOAD
 TAMO
TAMO
:: MORE INFORMATION
Citation:
D. Benjamin Gordon, Lena Nekludova, Scott McCallum and Ernest Fraenke
TAMO: a flexible, object-oriented framework for analyzing transcriptional regulation using DNA-sequence motifs.
Bioinformatics. 2005 Jul 15;21(14):3164-5.