ArrayCluster 1.0
:: DESCRIPTION
ArrayCluster is one of the significant challenges in gene expression analysis to find unknown subtypes of several diseases at the molecular levels. This task can be addressed by grouping gene expression patterns of the collected samples on the basis of a large number of genes. Application of commonly used clustering methods to such a dataset however are likely to fail due to over-learning, because the number of samples to be grouped is much smaller than the data dimension which is equal to the number of genes involved in the dataset. To overcome such difficulty, we developed a novel model-based clustering method, referred to as the mixed factors analysis.
::DEVELOPER
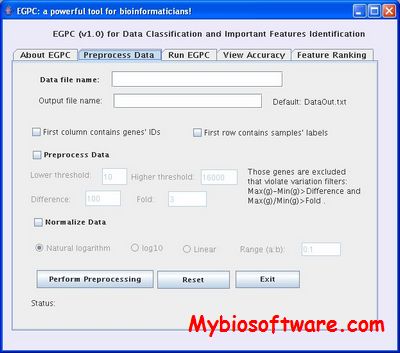
:: SCREENSHOTS

:: REQUIREMENTS
- Windows
:: DOWNLOAD

:: MORE INFORMATION
Citation
Bioinformatics. 2006 Jun 15;22(12):1538-9.
ArrayCluster: an analytic tool for clustering, data visualization and module finder on gene expression profiles.
Yoshida R, Higuchi T, Imoto S, Miyano S.