QTModel 0.70Beta
:: DESCRIPTION
QTModel is user-friendly computer software which packaged with modules for microarray data analysis, diallele design analysis and mixed model analysis.The mixed model module is developed for analyzing data from experimental designs with random factors. It is now available for commonly used randomized block design, randomized complete block design, latin square design, factorial design, multi-factor factorial design, nested design, and cross nested design etc. For fixed factors, pair-wised comparisons are done for all possible pairs of fixed effects of one factor. For random factors, some mixed linear model approaches, such as MINQUE, MIVQUE, REML and EM, will be used to estimate the variances of these random factors, and also unbiased prediction methods, such as BLUP, LUP and AUP, are used to predict the random effects of the random factors.
::DEVELOPER
ZJU-IBI
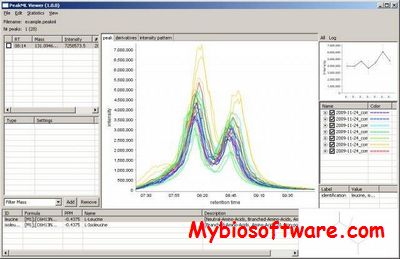
:: SCREENSHOTS

:: REQUIREMENTS
:: DOWNLOAD
 QTModel
QTModel
:: MORE INFORMATION
Citation
Funct Integr Genomics. 2009 Feb;9(1):59-66. Epub 2008 Sep 5.
Identifying differentially expressed genes in human acute leukemia and mouse brain microarray datasets utilizing QTModel.
Yang J, Zou Y, Zhu J.