CoStat 6.45
:: DESCRIPTION
CoStat is an easy-to-use program for data manipulation and statistical analysis.
::DEVELOPER
:: SCREENSHOTS

:: REQUIREMENTS
- Linux / Mac OsX / Windows
- Java
:: DOWNLOAD
:: MORE INFORMATION
:: DESCRIPTION
Pro-origami is a system for automatically generating protein structure cartoons. The cartoons are intended to make protein structure easy to interpret by laying out the secondary and super-secondary structure in two dimensions in a manner that makes the structure clear.
::DEVELOPER
IALab: Immersive Analytics Lab
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation
Bioinformatics. 2011 Dec 1;27(23):3315-6. doi: 10.1093/bioinformatics/btr575.
Automatic generation of protein structure cartoons with Pro-origami.
Stivala A1, Wybrow M, Wirth A, Whisstock JC, Stuckey PJ.
:: DESCRIPTION
iDNA-ABT is an advanced deep learning model that utilizes adaptive embedding based on bidirectional transformers for language understanding (BERT) together with a novel transductive information maximization (TIM) loss.
::DEVELOPER
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: MORE INFORMATION
Citation
Yu Y, He W, Jin J, Cui L, Zeng R, Wei L.
iDNA-ABT : advanced deep learning model for detecting DNA methylation with adaptive features and transductive information maximization.
Bioinformatics. 2021 Oct 2:btab677. doi: 10.1093/bioinformatics/btab677. Epub ahead of print. PMID: 34601568.
:: DESCRIPTION
BioAnnote is a desktop application is able to annotate biomedical texts by using different high-quality online resources, such as Medlineplus and Freebase.
::DEVELOPER
:: SCREENSHOTS
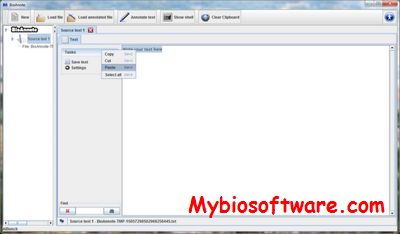
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation
Comput Methods Programs Biomed. 2013 Jul;111(1):139-47. doi: 10.1016/j.cmpb.2013.03.007.
BioAnnote: a software platform for annotating biomedical documents with application in medical learning environments.
López-Fernández H, Reboiro-Jato M, Glez-Peña D, Aparicio F, Gachet D, Buenaga M, Fdez-Riverola F.
:: DESCRIPTION
BioClass is a tool for biomedical text classification. Through it, a researcher can split a document set, directly related with a specific topic, into relevant or irrelevant documents. BioClass also supports several algorithms in order to increase the classification process efficiency and provides a set of powerful interfaces to analyse, filter and compare obtained results. In addition, all the operations than can be performed in BioClass are connected between them, so that the classification process is completely guided.
::DEVELOPER
:: SCREENSHOTS
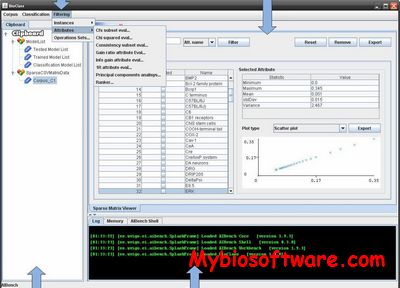
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
:: DESCRIPTION
pymzML is an extension to Python that offers
::DEVELOPER
CELLULAR AND MOLECULAR FUNCTIONS OF REACTIVE OXYGEN SPECIES
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation
Bald, T., Barth, J., Niehues, A., Specht, M., Hippler, M., and Fufezan, C. (2012)
pymzML – Python module for high throughput bioinformatics on mass spectrometry data,
Bioinformatics, doi: 10.1093/bioinformatics/bts066
:: DESCRIPTION
PIUMet is a network-based algorithm for integrative analysis of untargeted metabolomic data. It leverages known metabolic reactions and protein-protein interactions to analyze the ambiguous assignment of metabolomics features and identify disease-associated pathways and hidden components.
::DEVELOPER
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: DOWNLOAD
 NO
NO
:: MORE INFORMATION
Citation
Pirhaji L, Milani P, Leidl M, Curran T, Avila-Pacheco J, Clish CB, White FM, Saghatelian A, Fraenkel E.
Revealing disease-associated pathways by network integration of untargeted metabolomics.
Nat Methods. 2016 Sep;13(9):770-6. doi: 10.1038/nmeth.3940. Epub 2016 Aug 1. PMID: 27479327; PMCID: PMC5209295.
:: DESCRIPTION
DRUID combines IBD segments from a set of close relatives to reconstruct the IBD sharing profile of one of their ungenotyped ancestors. It uses this information to estimate relatedness between the ancestor and other more distant relatives.
::DEVELOPER
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation:
Ramstetter MD, Shenoy SA, Dyer TD, Lehman DM, Curran JE, Duggirala R, Blangero J, Mezey JG, Williams AL.
Inferring Identical-by-Descent Sharing of Sample Ancestors Promotes High-Resolution Relative Detection.
Am J Hum Genet. 2018 Jul 5;103(1):30-44. doi: 10.1016/j.ajhg.2018.05.008. Epub 2018 Jun 21. PMID: 29937093; PMCID: PMC6035284.
:: DESCRIPTION
PIVOT is an R-based platform that wraps open source transcriptome analysis packages with a uniform user interface and graphical data management that allows non-programmers to interactively explore transcriptomics data.
::DEVELOPER
:: SCREENSHOTS
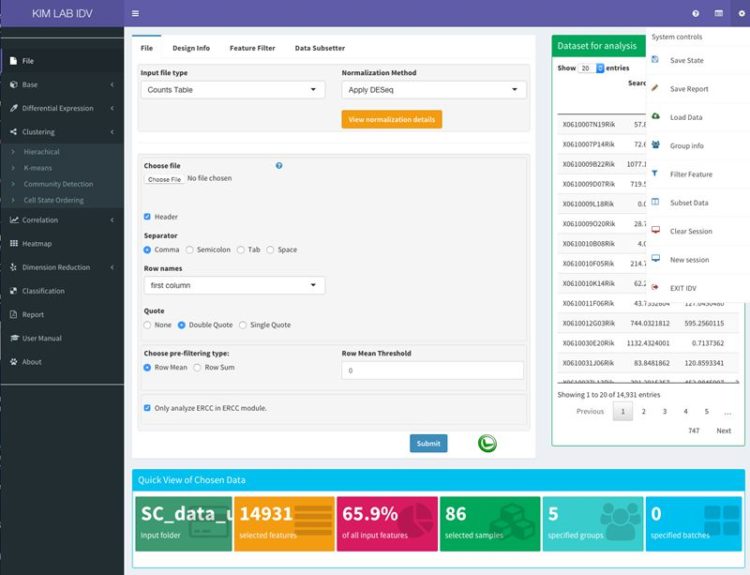
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation
Zhu Q, Fisher SA, Dueck H, Middleton S, Khaladkar M, Kim J.
PIVOT: platform for interactive analysis and visualization of transcriptomics data.
BMC Bioinformatics. 2018 Jan 5;19(1):6. doi: 10.1186/s12859-017-1994-0. PMID: 29304726; PMCID: PMC5756333.
:: DESCRIPTION
PESS (Protein Empirical Structure Space) is a software of sensitive protein fold recognition using an empirical structure space and 1NN.
::DEVELOPER
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation
Middleton SA, Illuminati J, Kim J.
Complete fold annotation of the human proteome using a novel structural feature space.
Sci Rep. 2017 Apr 13;7:46321. doi: 10.1038/srep46321. PMID: 28406174; PMCID: PMC5390313.