ELM / iELM 1.0
:: DESCRIPTION
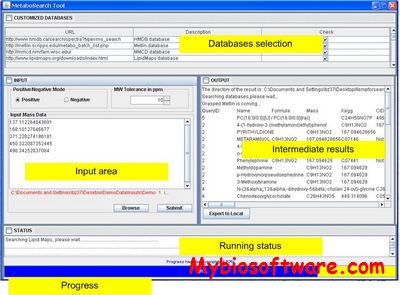
The ELM (Eukaryotic Linear Motif) resource provides the biological community with a comprehensive database of known experimentally validated motifs, and an exploratory tool to discover putative linear motifs in user-submitted protein sequences.
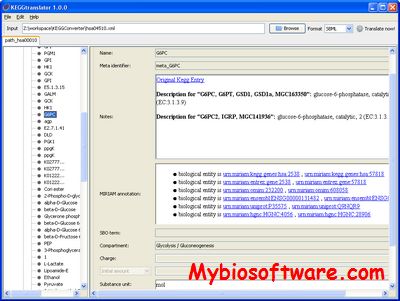
The iELM (interactions of Eukaryotic Linear Motif) web server provides a resource for predicting the function and positional interface for a subset of interactions mediated by short linear motifs (SLiMs).
::DEVELOPER
:: SCREENSHOTS

:: REQUIREMENTS
- Web Browser
:: DOWNLOAD
 NO
NO
:: MORE INFORMATION
Citation
Holger Dinkel et al.
ELM — the database of eukaryotic linear motifs
Nucl. Acids Res. (2012) 40 (D1): D242-D251.
iELM – a web server to explore short linear motif-mediated interactions.
Weatheritt RJ, Jehl P, Dinkel H, Gibson TJ. (2012).
Nucleic Acids Res. 2012 Jul