PRUNETREE 5.0
:: DESCRIPTION
Prunetree is a program for trimming taxa off phylogenies. You can make subtrees by either including (or excluding) a specific group of taxa. In addition, it also has options to arbitrarily transform internode branch lengths (BL) following a speciational model (all BL = 1) or following Pagel?s method (all taxa aligned and all internode BL = 1).
::DEVELOPER
:: SCREENSHOTS
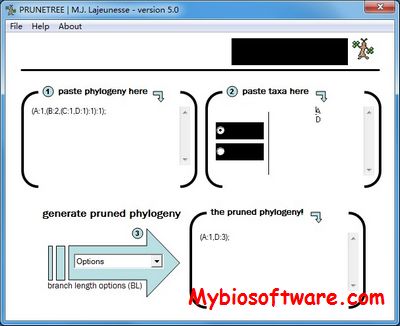
:: REQUIREMENTS
- Windows
:: DOWNLOAD

:: MORE INFORMATION