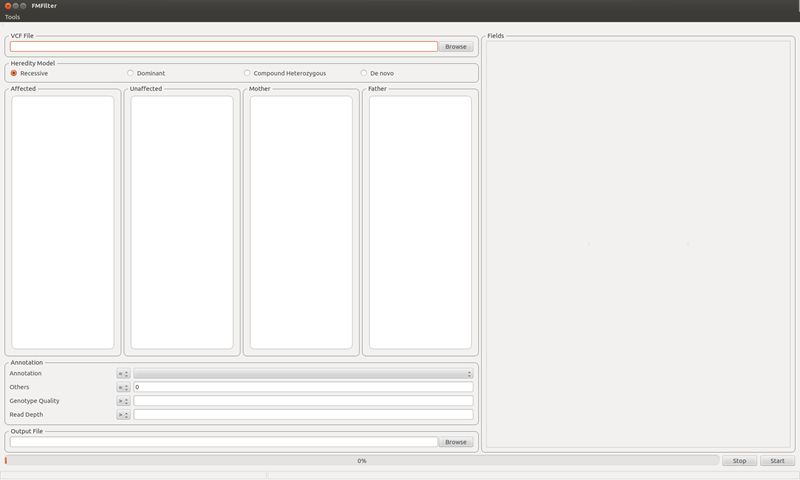
PURE
:: DESCRIPTION
PURE is a PubMed article recommendation system based on content-based filtering. PURE has a web interface by which users can add and/or delete their preferred articles. Once articles are registered, PURE then performs model-based clustering of the preferred articles and recommends highly-rated articles by prediction using the trained model. PURE updates PubMed articles and reports recommendation by email on daily-base.
::DEVELOPER
Bio-Knowledge Engineering Research Laboratory
:: SCREENSHOTS
N/A
:: REQUIREMENTS
- WIndows/Linux/MacOsX
- Perl
- MySQL
- PHP
- APACHE
:: DOWNLOAD

:: MORE INFORMATION
Citation:
Genome Inform. 2007;18:267-76.
PURE: a PubMed article recommendation system based on content-based filtering.
Yoneya T, Mamitsuka H.