QuasiMotiFinder v4
:: DESCRIPTION
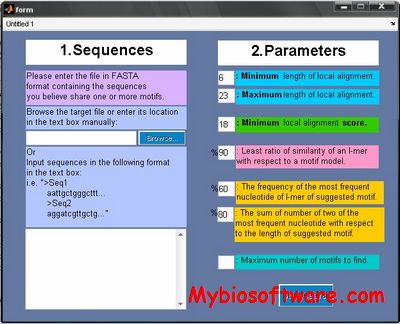
QuasiMotiFinder is a server for the identification of signatures and signature-like patterns in protein sequences
::DEVELOPER
:: SCREENSHOTS
N/A
:: REQUIREMENTS
- Web browser
:: DOWNLOAD
 NO
NO
:: MORE INFORMATION
Citation:
Nucleic Acids Res. 2005 Jul 1;33(Web Server issue):W255-61.
QuasiMotiFinder: protein annotation by searching for evolutionarily conserved motif-like patterns.
Gutman R, Berezin C, Wollman R, Rosenberg Y, Ben-Tal N.