DISCO 1.0
:: DESCRIPTION
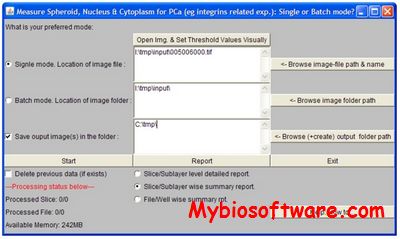
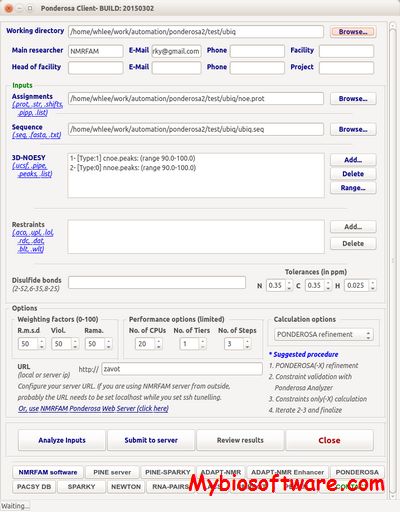
DISCO is software to perform structure determination of protein homo-oligomers with cyclic symmetry.DISCO computes oligomeric protein structures using geometric constraints derived from RDCs and intermolecular distance restraints such as NOEs or disulfide bonds. When a reliable subunit structure can be calculated from intramolecular restraints, DISCO guarantees that all satisfying oligomer structures will be discovered, yet can run in minutes to hours on only a single desktop-class computer.
::DEVELOPER
Donald Lab at Duke University
:: SCREENSHOTS
N/A
:: REQUIREMENTS
- Windows / Linux / MacOSX
- Java
:: DOWNLOAD

:: MORE INFORMATION
Citation
Jeffrey W. Martin, Anthony K. Yan, Chris Bailey-Kellogg, Pei Zhou, and Bruce R. Donald.
A graphical method for analyzing distance restraints using residual dipolar couplings for structure determination of symmetric protein homo-oligomers.
Protein Science, 20(6):970-985, 2011.