MulteeSum
:: DESCRIPTION
MulteeSum is a visualization system that supports inspection and curation of data sets showing gene expression over time, in conjunction with the spatial location of the cells where the genes are expressed — it is the first tool to support comparisons across multiple such datasets.
::DEVELOPER
:: SCREENSHOTS
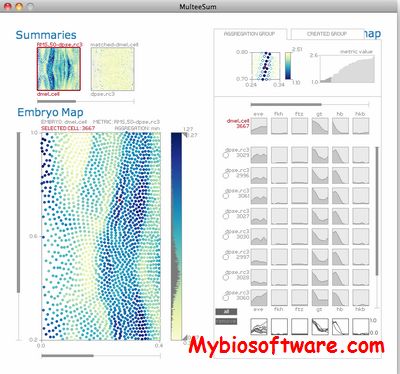
:: REQUIREMENTS
- Linux/ Windows/ MacOsX
:: DOWNLOAD

:: MORE INFORMATION
Citation:
IEEE Trans Vis Comput Graph. 2010 Nov-Dec;16(6):908-17. doi: 10.1109/TVCG.2010.137.
MulteeSum: a tool for comparative spatial and temporal gene expression data.
Meyer M, Munzner T, DePace A, Pfister H.






