samql v1.6
:: DESCRIPTION
SamQL is a structured query language and filtering tool for the SAM/BAM file format.
::DEVELOPER
:: SCREENSHOTS
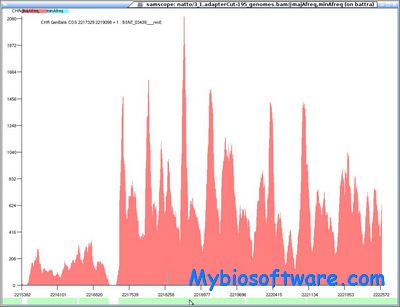
N/A
:: REQUIREMENTS
- Linux
:: DOWNLOAD

:: MORE INFORMATION
Citation
Lee CT, Maragkakis M.
SamQL: a structured query language and filtering tool for the SAM/BAM file format.
BMC Bioinformatics. 2021 Oct 2;22(1):474. doi: 10.1186/s12859-021-04390-3. PMID: 34600480; PMCID: PMC8487582.