sig 1.0
:: DESCRIPTION
sig is a program to search multiple occurences of multiple motifs in a set of sequences.
::DEVELOPER
Eric Deveaud <edeveaud@pasteur.fr>
:: SCREENSHOTS
N/A
:: REQUIREMENTS
- Linux
:: DOWNLOAD
:: MORE INFORMATION
:: DESCRIPTION
Mauve is a system for efficiently constructing multiple genome alignments in the presence of large-scale evolutionary events such as rearrangement and inversion. Multiple genome alignment provides a basis for research into comparative genomics and the study of evolutionary dynamics on a new scale. Aligning whole genomes is a fundamentally different problem than aligning short sequences.
Mauve has been developed with the idea that a multiple genome aligner should require only modest computational resources. It employs algorithmic techniques that scale well in the amount of sequence being aligned. For example, a pair of Y. pestis genomes can be aligned in under a minute, while a group of 9 divergent Enterobacterial genomes can be aligned in a few hours.
::DEVELOPER
:: SCREENSHOTS

:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation
Aaron C.E. Darling, Bob Mau, Frederick R. Blatter, and Nicole T. Perna. 2004.
Mauve: multiple alignment of conserved genomic sequence with rearrangements.
Genome Research. 14(7):1394-1403.
:: DESCRIPTION
PMFastR is an algorithm which iteratively uses a sequence-structure alignment procedure to build a multiple RNA structure alignment
::DEVELOPER
UCF Computational Biology and Bioinformatics Group
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation
IEEE/ACM Trans Comput Biol Bioinform. 2011 Apr 29. [Epub ahead of print]
A Memory Efficient Method for Structure-Based RNA Multiple Alignment.
Deblasio D, Bruand J, Zhang S.
:: DESCRIPTION
MIL(multiple-instance learning) is a novel algorithm, which breaks each DNA sequence into multiple overlapping subsequences and models each subsequence separately, therefore implicitly takes into consideration binding site locations, resulting in both higher accuracy and better interpretability of the models.
::DEVELOPER
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation
Computational modeling of in vivo and in vitro protein-DNA interactions by multiple instance learning.
Gao Z, Ruan J.
Bioinformatics. 2017 Jul 15;33(14):2097-2105. doi: 10.1093/bioinformatics/btx115.
:: DESCRIPTION
SARA-Coffee is a structure based multiple RNA aligner. This is a new algorithm that joins the pairwise RNA structure alignments performed by SARA with the multiple sequence T-Coffee framework. SARA-Coffee is part of the T-Coffee distribution.
::DEVELOPER
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation:
SARA-Coffee web server, a tool for the computation of RNA sequence and structure multiple alignments.
Di Tommaso P, Bussotti G, Kemena C, Capriotti E, Chatzou M, Prieto P, Notredame C.
Nucleic Acids Res. 2014 Jul;42(Web Server issue):W356-60. doi: 10.1093/nar/gku459.
:: DESCRIPTION
SMAT is an R package for performing the Scaled Multiple-phenotype Association Test in cohort or case-control designs to assess common effect of a single nucleotide polymorphism (SNP) on multiple (positively correlated) continuous outcomes measuring the same underlying trait.
::DEVELOPER
Xihong Lin’s Group, Harvard School of Public Health
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: MORE INFORMATION
Citation
Genome-wide Association Analysis for Multiple Continuous Secondary Phenotypes.
Schifano ED, Li L, Christiani DC, Lin X.
Am J Hum Genet. 2013 May 2;92(5):744-59.
:: DESCRIPTION
Mugsy is a multiple whole genome aligner. Mugsy uses Nucmer for pairwise alignment, a custom graph based segmentation procedure for identifying collinear regions, and the segment-based progressive multiple alignment strategy from Seqan::TCoffee. Mugsy accepts draft genomes in the form of multi-FASTA files and does not require a reference genome.
::DEVELOPER
Samuel V. Angiuoli , Dr. Steven Salzberg.
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation
Angiuoli SV and Salzberg SL.
Mugsy: Fast multiple alignment of closely related whole genomes.
Bioinformatics 2011 27(3):334-4
:: DESCRIPTION
The Tumorscape portal facilitates the use and understanding of high resolution copy number data amassed from multiple cancer types. It supports gene-level analysis, analysis by cancer type, and the downloading/browsing of data.
::DEVELOPER
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: DOWNLOAD
 NO
NO
:: MORE INFORMATION
Citation
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M. et al.
The landscape of somatic copy-number alteration across human cancers.
Nature. 2010;463:899–905. doi: 10.1038/nature08822.
:: DESCRIPTION
sStu (Sequences studio) is a java package to perform multiple kinds of sequence alignments.
::DEVELOPER
:: SCREENSHOTS
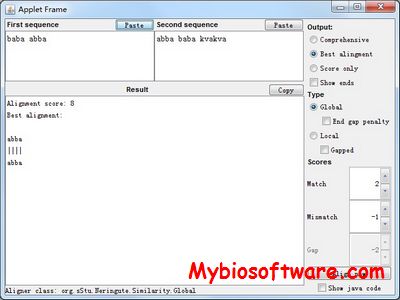
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
:: DESCRIPTION
MGRA is a software for reconstruction of phylogenetic trees as well as ancestral genomes and applied it to study the rearrangement history of seven mammalian genomes: human, chimpanzee, macaque, mouse, rat, dog, and opossum. MGRA relies on the new notion of the multiple breakpoint graphs to overcome some limitations of the existing approaches to ancestral genome reconstructions. In particular, we applied \MGRA to analyze the primate–rodent–carnivore controversy in mammalian phylogeny, i.e., the alternative between the primate–rodent and primate–carnivore clades. MGRA provided the rearrangement-based evidence, albeit limited, for the primate–carnivore clade as opposed to the currently favored primate–rodent clade.
::DEVELOPER
Max Alekseyev and Pavel A. Pevzner
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation:
Max Alekseyev, Pavel Pevzner
“Breakpoint Graphs and Ancestral Genome Reconstructions.”
Genome Res. 2009 May;19(5):943-57. Epub 2009 Feb 13.