Sequencher 5.4.6
:: DESCRIPTION
Sequencher is the industry standard software for DNA sequence analysis. It works with all automated sequencers and is widely known for its lightning-fast contig assembly, short learning curve, user-friendly editing tools, and superb technical support. First released almost 15 years ago, Sequencher is currently used for sequence analysis tasks in every major genomic and pharmaceutical company as well as numerous academic and government labs in over 40 countries around the world. Life Science researchers use Sequencher for many diverse DNA sequence analysis applications including de novo gene sequencing, mutation detection, forensic human identification, systematics, and more.
::DEVELOPER
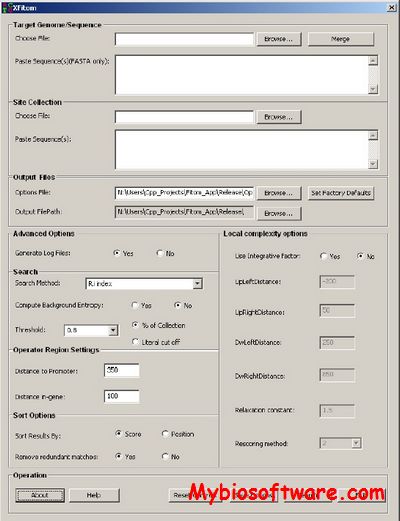
:: SCREENSHOTS

:: REQUIREMENTS
- Windows / MacOsX
:: DOWNLOAD

:: MORE INFORMATION