ChIPpeakAnno 3.24.21
:: DESCRIPTION
ChIPpeakAnno is a Bioconductor package within the statistical programming environment R to facilitate batch annotation of enriched peaks identified from ChIP-seq, ChIP-chip, cap analysis of gene expression (CAGE) or any experiments resulting in a large number of enriched genomic regions.
::DEVELOPER
Program in Gene Function and Expression@umassmed
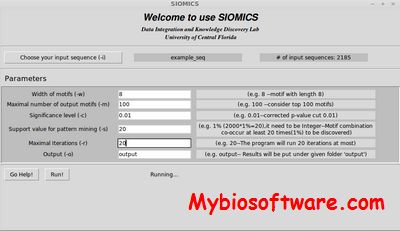
:: SCREENSHOTS
N/A
:: REQUIREMENTS
- Windows/Linux/MacOsX
- BioConductor
- R package
:: DOWNLOAD

:: MORE INFORMATION
Citation:
BMC Bioinformatics. 2010 May 11;11:237. doi: 10.1186/1471-2105-11-237.
ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data.
Zhu LJ, Gazin C, Lawson ND, Pagès H, Lin SM, Lapointe DS, Green MR.