Farsight 0.4.4
:: DESCRIPTION
The goal of the FARSIGHT project is to develop and disseminate a next-generation toolkit of image analysis methods to enable quantitative studies of complex & dynamic tissue microenvironments that are imaged by modern optical microscopes. Examples of such microenvironments include brain tissue, stem cell niches, developing embryonic tissue, immune system components, and tumors. Progress in mapping these microenvironments is much too slow compared to the need. Our knowledge of these systems has been painstakingly “pieced together” from large numbers of fixed, 2-D images of specimens revealing a small fraction of the molecular ‘players’ involved. The goal of this project is to help accelerate progress by: (i) harnessing the power of modern microscopy to help see the microenvironments in a much more detailed, direct, and comprehensive manner; and (ii) computational tools to analyze the multi-dimensional data produced by these microscopes.
::DEVELOPER
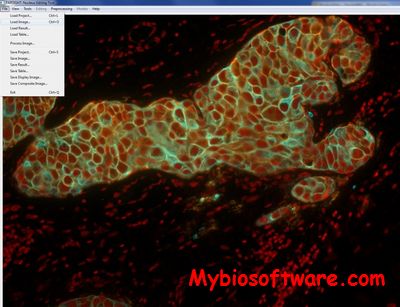
:: SCREENSHOTS
:: REQUIREMENTS
- Windows / MacOSX /Linux
:: DOWNLOAD

:: MORE INFORMATION
Citaition
The FARSIGHT trace editor: an open source tool for 3-D inspection and efficient pattern analysis aided editing of automated neuronal reconstructions.
Luisi J, Narayanaswamy A, Galbreath Z, Roysam B.
Neuroinformatics. 2011 Sep;9(2-3):305-15. doi: 10.1007/s12021-011-9115-0.