sStu 1.1
:: DESCRIPTION
sStu (Sequences studio) is a java package to perform multiple kinds of sequence alignments.
::DEVELOPER
:: SCREENSHOTS
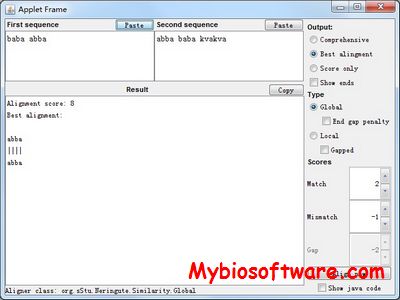
:: REQUIREMENTS
- Windows / Linux / MacOsX
- Java
:: DOWNLOAD

:: MORE INFORMATION
:: DESCRIPTION
sStu (Sequences studio) is a java package to perform multiple kinds of sequence alignments.
::DEVELOPER
:: SCREENSHOTS
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
:: DESCRIPTION
WHAM (WISCONSIN’S HIGH-THROUGHPUT ALIGNMENT METHOD) is a high-throughput sequence alignment tool developed at University of Wisconsin-Madison. It aligns short DNA sequences (reads) to the whole human genome at a rate of over 1500 million 60bps reads per hour, which is one to two orders of magnitudes faster than the leading state-of-the-art techniques.
::DEVELOPER
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation:
WHAM: A High-throughput Sequence Alignment Method,
Y. Li, A. Terrel and J. M. Patel,
SIGMOD 2011, Athens, Greece
:: DESCRIPTION
SEQMOL (“sequences & molecules”) is an integrated sequence alignment and PDB structure analysis utility. It can be used to align multiple protein and DNA sequences, compute evolutionary attributes of multiple sequence alignments (such as sequence conservation, hydrophobicity conservation, conformational flexibility conservation, physical covariation, protein-protein interface, protein-RNA interface and protein-DNA interface propensity, and conservations thereof) and to map these features onto PDB files.
::DEVELOPER
:: SCREENSHOTS


:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
:: DESCRIPTION
ImOSM (Imbed One Step Mutations into an alignment) is a program to Imbed model violation as One Step Mutations into a sequence alignment. This is a useful tool to study the robustness in phylogeny inference.
::DEVELOPER
the Center of Integrative Bioinformatics Vienna (CIBIV) headed by Arndt von Haeseler.
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation:
Minh Anh Thi Nguyen, Tanja Gesell, and Arndt von Haeseler.
ImOSM: Intermittent evolution and robustness of phylogenetic methods.
Mol Biol Evol (2011) doi: 10.1093/molbev/msr220
:: DESCRIPTION
ISAS (Imagenix Sequence Alignment System )is a software for DNA Sequence Alignment, Array Probe Design, or Genomics Research. ISAS can be run on a single computer, and produce the same results, in less time than the standard sequence alignment software does when running on a big cluster of computers
::DEVELOPER
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation:
Bioinformatics. 2011 Jan 15;27(2):272-4. Epub 2010 Nov 12.
The uniqueome: a mappability resource for short-tag sequencing.
Koehler R, Issac H, Cloonan N, Grimmond SM.
:: DESCRIPTION
CBESWdemonstrates how the PlayStation® 3, powered by the Cell Broadband Engine, can be used as a computational platform to accelerate the Smith-Waterman algorithm. It also demonstrates that the PlayStation® 3 console can be used as an efficient low cost computational platform for high performance sequence alignment applications.For large datasets, our implementation on the PlayStation® 3 provides a significant improvement in running time compared to other implementations such as SSEARCH, Striped Smith-Waterman and CUDA. CBESW achieves a peak performance of up to 3.646 GCUPS. This performance is about 30x faster than the straightforward C-implementation in SSEARCH. It is also 1.64x faster than the highly optimized striped SW implementation on an Intel processor and 3x times faster than a CUDA implementation on an Nvidia GeForce 8800GTX.
::DEVELOPER
Parallel and Distributed Architectures
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation
Adrianto Wirawan, Chee Keong Kwoh, Tri Hieu Nim, Bertil Schmidt:
“CBESW: Sequence Alignment on the Playstation 3“.
BMC Bioinformatics, 2008, 9:377
:: DESCRIPTION
CINEMA 5 (Colour INteractive Editor for Multiple Alignments) is an interactive visual tool for the interpretation and manipulation of protein and DNA sequences. CINEMA 5 allows the user to interactively construct and edit and visualise multiple sequence alignments.
::DEVELOPER
:: SCREENSHOTS

:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation
Bioinformatics. 2002 Oct;18(10):1402-3.
CINEMA-MX: a modular multiple alignment editor.
Lord PW, Selley JN, Attwood TK.
:: DESCRIPTION
GS-Aligner that uses bit-level operations was developed for aligning genomic sequences. GS-Aligner is efficient in terms of both time and space for aligning two very long genomic sequences and for identifying genomic rearrangements such as translocations and inversions. It is suitable for aligning fairly divergent sequences such as human and mouse genomic sequences. It consists of several efficient components: bit-level coding, search for matching segments between the two sequences as alignment anchors, longest increasing subsequence (LIS), and optimal local alignment. Efforts have been made to reduce the execution time of the program to make it truly practical for aligning very long sequences. Empirical tests suggest that for relatively divergent sequences such as sequences from different mammalian orders or from a mammal and a nonmammalian vertebrate GS-Aligner performs better than existing methods.
::DEVELOPER
Chun-Chieh Shih and Wen-Hsiung Li
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation:
GS-Aligner: A Novel Tool for Aligning Genomic Sequences Using Bit-Level Operations
Chun-Chieh Shih and Wen-Hsiung Li
Mol Biol Evol (2003) 20 (8): 1299-1309.
:: DESCRIPTION
GoCore (Grouping of Conserved regions)is a free bioinformatics tool for protein sequence alignment and analysis. It operates as an Excel add-in, creating additional menu items that perform the bioinformatics analyses and uses Excel’s convenient user interface to provide useful visualizations. Anybody who can use Excel will find it easy to use GoCore.
::DEVELOPER
:: SCREENSHOTS

:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation
Gilchrist RB, Ritter LJ, Cranfield M, Jeffery LA, Amato F, Scott SJ, Myllymaa S, Kaivo-Oja N, Lankinen H, Mottershead DG, Groome NP, Ritvos O.
Immunoneutralization of growth differentiation factor 9 reveals it partially accounts for mouse oocyte mitogenic activity
Biology of Reproduction 2004 Sep; 71(3): 732-9
:: DESCRIPTION
probA – calculates the partiton function of a pairwise global alignment. The partition function is then used to compute the probability of every possible match (or mismatch) between the two sequences. The partition function is also employed for stochastic backtracking which generates a correctly weighted ensemble of optimal and suboptimal alignments.
::DEVELOPER
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
Citation
Bioinformatics. 2002;18 Suppl 2:S153-60.
Stochastic pairwise alignments.
Mückstein U, Hofacker IL, Stadler PF.