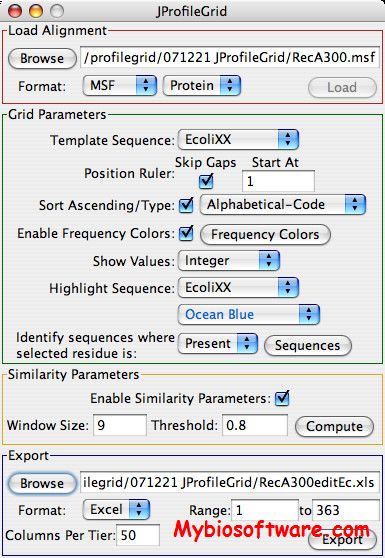
Multi-Relief
:: DESCRIPTION
multi-RELIEF is a novel approach for identifying specificity residues that is based on RELIEF, a state-of-the-art Machine-Learning technique for feature weighting.
::DEVELOPER
:: SCREENSHOTS
N/A
:: REQUIREMENTS
- Linux/Windows
- MatLab
:: DOWNLOAD

:: MORE INFORMATION
Citation
K. Ye, A. Feenstra, A.Ijzerman, J. Heringa, E. Marchiori.
Multi-RELIEF: a method to recognize specificity determining residues from multiple sequence alignments using a Machine Learning approach for feature weighting.
Bioinformatics, 24(1), pp. 18-25, 2008.