ProteoSAFe is a Proteomics Environment which is Scalable in utilizing distributed computing, Accessible via reconfigurable, easy-to-learn user interfaces, and Flexible in tool chaining.
Inspect is a general purpose database search algorithm, with an emphasis on efficiently and confidently identifying modified peptides. It includes special scoring models for phosphorylation which allow for increased accuracy. In addition, Inspect implements the MS-Alignment algorithm for discovery of unanticipated modifications in blind mode.
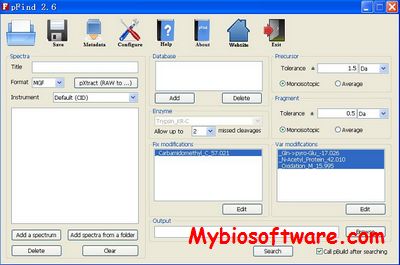
pFind Studio is a computational solution for mass spectrometry-based proteomics. pFind Studio includes pFind, pBuild, pLabel, pXtract, pParse and pScan
pXtract creates .DTA .MGF and .MS2 input files directly from Thermo Scientific .raw LC-MS/MS data files.
pParse is a software dedicated to recalibrate the monoisotopic of precursors in MS/MS spectra datasets assigned by mass spectrometry
pFind is a search engine for peptide and protein identification via tandem mass spectrometry.
pNovo+ is a de novo peptide sequencing algorithm using complementary HCD and ETD tandem mass spectra.
pBuild is a tool that can compare several search engines’ results and combine them together. The latest version, pBuild v2.0, can process the search results of pFind, SEQUEST and Mascot.
pQuant is the software for quantitative proteomics, evaluates the accuracy of caluculated peptide and protein ratios.
pLabel is a spectra labeling tool that can visualize the global- and local-view peptide-spectrum matches, given the results of pFind or any other search engines. pLabel can label both CID and ETD spectra, and implement the manual de novo sequencing.
pLink is a software dedicated for the analysis of chemically cross-linked proteins or protein complexes using mass spectrometry.
pScan is a flexible tool that helps biologists to preprocess protein sequence databases in proteomics research.
pCluster is a software tool aiming at detecting protein modifications independent of sequence databases by tandem mass spectral clustering.
pMatch is a spectral library search tool, which is deliberately designed for the open search mode.
::DEVELOPER
Bioinformatics group, Institute of Computing Technology, Chinese Academy of Sciences
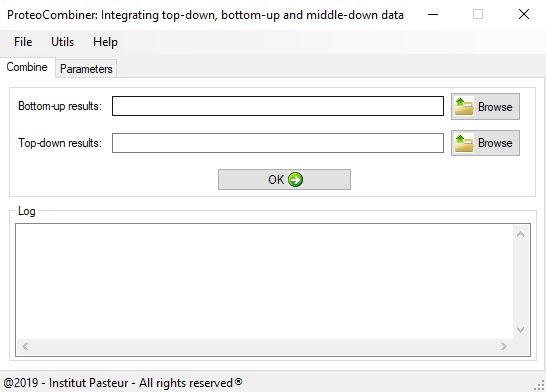
ProteoCombiner capitalizes on the data arising from different experiments and proteomics search engines and presents the results in a user-friendly manner. Our tool also provides a rapid and easy visualization, manual validation and comparison of the identified proteoform sequences, including post-translational modifications (PTM) characterization.
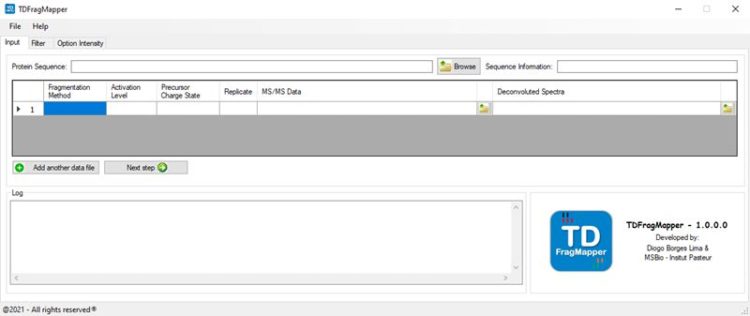
TDFragMapper can deal with multiple fragmentation methods and can rapidly highlight the experimental fragmentation parameters that are critical to the characterization of intact proteins of various size using top-down proteomics.
ProHits is an open source software package designed to help scientists store, search and analyze mass spectrometry data, in particular for protein-protein interaction experiments.
Philosopher provides easy access to third-party tools and custom algorithms for proteomics analysis, from database searching to functional protein reports. Philosopher is also well-equipped for open search analysis, providing extended versions of PeptideProphet and ProteinProphet for peptide validation and protein inference.
MSFragger is an ultrafast database search tool that uses a fragment ion indexing method to rapidly perform spectra similarity comparisons.
FragPipe is a Java Graphical User Interface (GUI) for a suite of computational tools enabling comprehensive analysis of mass spectrometry-based proteomics data. It is powered by MSFragger – an ultrafast proteomic search engine suitable for both conventional and “open” (wide precursor mass tolerance) peptide identification.
DIA-Umpire is an open source Java program for computational analysis of data independent acquisition (DIA) mass spectrometry-based proteomics data. It enables untargeted peptide and protein identification and quantitation using DIA data, and also incorporates targeted extraction to reduce the number of cases of missing quantitation.