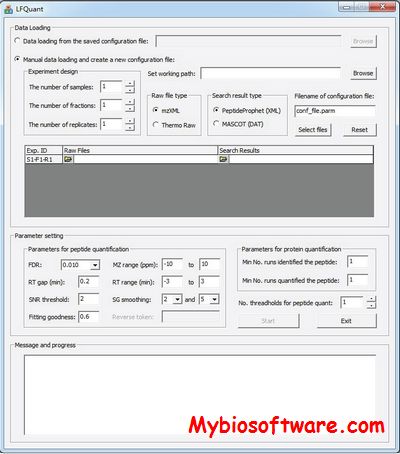
LFQuant is a new analysis tool for label-free LC-MS/MS quantitative proteomics data. It is compatible with high-resolution mass spectrometers (Thermo RAW data) and two popular database search engines (SEQUEST and MASCOT) with target-decoy search strategy.
aLFQ is a bioinformatics tool which supports the commonly used absolute label-free protein abundance estimation methods (TopN, iBAQ, APEX, NSAF and SCAMPI) for LC-MS/MS proteomics data, together with validation algorithms enabling automated data analysis and error estimation.
OpenMS is an open-source software C++ library for LC/MS data management and analyses. It offers an infrastructure for the development of mass spectrometry related software.
pyOpenMS provides Python-bindings for the C++ OpenMS mass spectrometric algorithm library, allowing researchers to directly access algorithms and data structures available in C++ from the interactive Python environment. pyOpenMS thus provides access to a feature-rich, open-source algorithm library for mass-spectrometry based proteomics analysis, giving the user functionality ranging from file access (mzXML, mzML, TraML, mzIdentML among others), basic signal processing (smoothing, filtering, de-isotoping and peak-picking) and complex data analysis (including label-free, SILAC, iTRAQ and SWATH analysis tools).
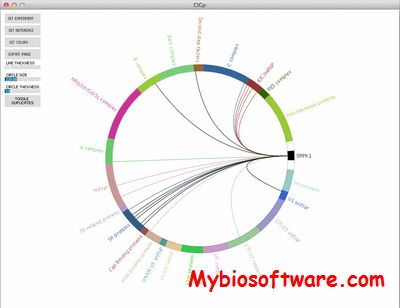
CIG-P is a higher order visualization tool for AP-MS which generates intuitive circular diagrams for visually appealing final representation of AP-MS data.
protViz is an R package that helps with quality checks, vizualizations and analysis of mass spectrometry data, coming from proteomics experiments. The package is developed, tested and used at the Functional Genomics Center Zurich. We use this package mainly for prototyping, teaching, and having fun with proteomics data. But it can also be used to do solid data analysis for small scale data sets.
EBprot is a novel probabilistic framework that directly models the peptide-protein hierarchy and rewards the proteins with reproducible evidence of DE over multiple peptides.
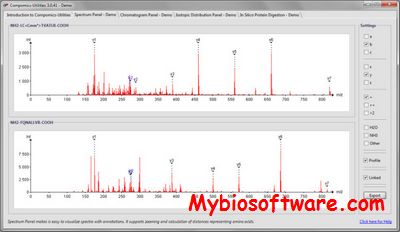
Compomics-utilities is an open-source support library for computational proteomics. The library contains a broad set of features required for reading, parsing, and analyzing proteomics data. compomics-utilities is already used by a long list of existing software, ensuring library stability and continued support and development.
Pyteomics is a collection of lightweight and handy tools for Python that help to handle various sorts of proteomics data. Pyteomics provides a growing set of modules to facilitate the most common tasks in proteomics data analysis
specL provides a function for generating spectra libraries which can be used for MRM SRM MS workflows in proteomics. The package provides a BiblioSpec reader, a function which can add the protein information using a FASTA formatted amino acid file, and an export method for using the created library in the Spectronaut software.