BiblioSpec 2.0
:: DESCRIPTION
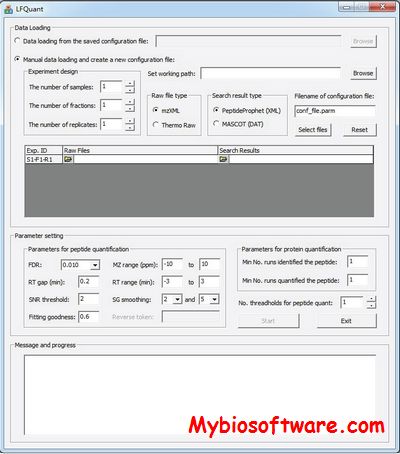
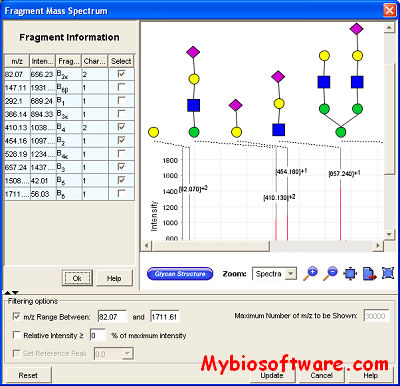
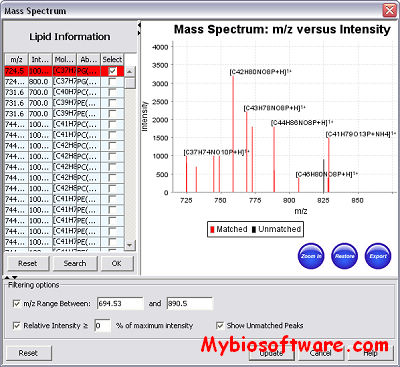
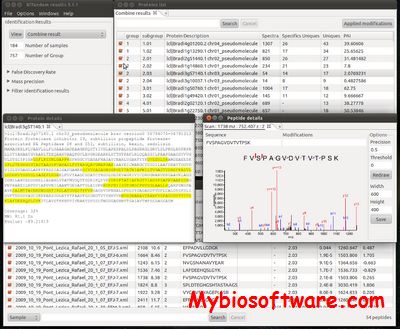
BiblioSpec enables the identification of peptides from tandem mass spectra by searching against a database of previously identified spectra.
::DEVELOPER
:: SCREENSHOTS
N/A
:: REQUIREMENTS
- Linux / Windows
:: DOWNLOAD

:: MORE INFORMATION
Citation
Using BiblioSpec for creating and searching tandem MS peptide libraries.
Frewen B, MacCoss MJ.
Curr Protoc Bioinformatics. 2007 Dec;Chapter 13:Unit 13.7.