AMEN (Annotation, Mapping, Expression and Network) is a stand-alone, unified suite of tools to manage, explore and combine biological multifaceted high-throughput data such as annotation, chromosomal location, expression and interaction data.
CDAO provides extensive conceptual coverage of commonly used concepts in evolutionary analyses in a single framework. It is developed in collaboration with a group of domain scientists specialising in phylogenetic analysis software, the EvoInfo working group (http://evoinfo.nescent.org) . CDAO is used in a number of projects, including nexML, Nexplorer3, TreeBASE, phyloXML.
QTModel is user-friendly computer software which packaged with modules for microarray data analysis, diallele design analysis and mixed model analysis.The mixed model module is developed for analyzing data from experimental designs with random factors. It is now available for commonly used randomized block design, randomized complete block design, latin square design, factorial design, multi-factor factorial design, nested design, and cross nested design etc. For fixed factors, pair-wised comparisons are done for all possible pairs of fixed effects of one factor. For random factors, some mixed linear model approaches, such as MINQUE, MIVQUE, REML and EM, will be used to estimate the variances of these random factors, and also unbiased prediction methods, such as BLUP, LUP and AUP, are used to predict the random effects of the random factors.
S-MART manages your RNA-Seq and ChIP-Seq data. It also produces many different plots to visualize your data.S-MART is an intuitive and lightweight tool, performing several tasks that are usually required during the analysis of mapped RNA-Seq and ChIP-Seq reads, including data selection and data visualization.
Philosopher provides easy access to third-party tools and custom algorithms for proteomics analysis, from database searching to functional protein reports. Philosopher is also well-equipped for open search analysis, providing extended versions of PeptideProphet and ProteinProphet for peptide validation and protein inference.
mzMatch is a modular, open source and platform independent data processing pipeline for metabolomics LC/MS data written in the Java language. It was designed to provide small tools for the common processing tasks for LC/MS data. The mzMatch environment was based entirely on the PeakML file format and core library, which provides a common framework for all the tools.
protViz is an R package that helps with quality checks, vizualizations and analysis of mass spectrometry data, coming from proteomics experiments. The package is developed, tested and used at the Functional Genomics Center Zurich. We use this package mainly for prototyping, teaching, and having fun with proteomics data. But it can also be used to do solid data analysis for small scale data sets.
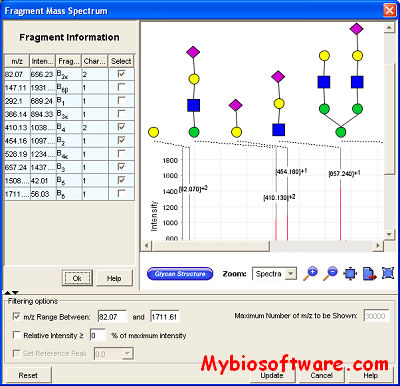
SimGlycan® predicts the structure of glycans and glycopeptides using mass spectrometry data. SimGlycan® accepts the experimental MS/MS data generated by a mass spectrometer, matches them with its own database of theoretical fragments and generates a list of probable candidate structures.
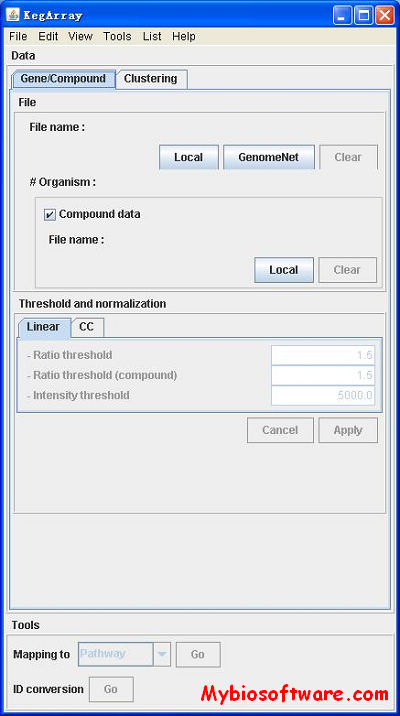
KegArray is a Java application that provides an environment for analyzing both transcriptome data (gene expression profiles) and metabolome data (compound profiles). Tightly integrated with the KEGG database, KegArray enables you to easily map those data to KEGG resources including PATHWAY, BRITE and genome maps.