AlignExIn ( Align Exon Intron) is a handy and useful utility built in order to display the alignment of exons. The program displays the exon painted in red and the alignments painted in blue by default, but it is possible to change the colors.All you have to do is select the input files (fasta and exon) and the program will do the rest.
NoFold is an approach for characterizing and clustering RNA secondary structures without computational folding or alignment. It works by mapping each RNA sequence of interest to a structural feature space, where each coordinate within the space corresponds to the probabilistic similarity of the sequence to an empirically defined structure model (e.g. Rfam family covariance models).
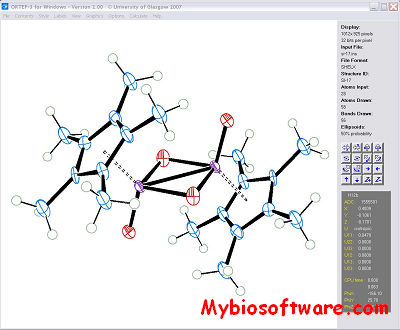
ORTEP-III is Oak Ridge Thermal Ellipsoid Plot Program for crystal structure illustrations.Ball-and-stick type illustrations of a quality suitable for publication are produced with either spheres or thermal-motion probability ellipsoids, derived from anisotropic temperature factor parameters, on the atomic sites. The program also produces stereoscopic pairs of illustrations which aid in the visualization of complex arrangements of atoms and their correlated thermal motion patterns.
Ortep-3 for Windows developed by Dr. Louis Farrugia is a MS-Windows version of the current release of ORTEP-III
Michael N. Burnett and Carroll K. Johnson,
ORTEP-III: Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustrations,
Oak Ridge National Laboratory Report ORNL-6895, 1996.
SMOG is a versatile software package for generating structure-based models.SMOG 2 is a downloadable software package that reads user-designated structural information and user-defined energy definitions, in order to produce the files necessary to use SBMs with high performance molecular dynamics packages: GROMACS and NAMD.
de Oliveira AB Jr, Contessoto VG, Hassan A, Byju S, Wang A, Wang Y, Dodero-Rojas E, Mohanty U, Noel JK, Onuchic JN, Whitford PC. SMOG 2 and OpenSMOG: Extending the limits of structure-based models.
Protein Sci. 2021 Oct 16. doi: 10.1002/pro.4209. Epub ahead of print. PMID: 34655449.
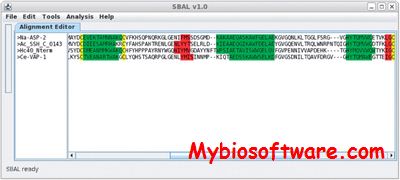
SBAL is intended for multiple protein sequence alignments guided by secondary structure elements. The program provides automatic and semi-automatic alignment features, and also possesses manual editing capabilities.
NASP is a computer program that will allow predict the most evolutionarily conserved secondary structures evident within a set of aligned nucleic acid sequences. It will progressively identify all of the most probable secondary structures that display some degree of sequence conservation between sequences in an analysed alignment.
LTR_STRUC is a new data-mining program that scans nucleotide sequence files for LTR retrotransposons and analyzes any resulting hits. Input files can be in fasta or NCBI flat file format.
DISCO is software to perform structure determination of protein homo-oligomers with cyclic symmetry.DISCO computes oligomeric protein structures using geometric constraints derived from RDCs and intermolecular distance restraints such as NOEs or disulfide bonds. When a reliable subunit structure can be calculated from intramolecular restraints, DISCO guarantees that all satisfying oligomer structures will be discovered, yet can run in minutes to hours on only a single desktop-class computer.
QRNAS (Quick Refinement of Nucleic Acid Structures) is an extension of the AMBER simulation method with additional terms associated with explicit hydrogen bonds, co-planarity base pairs, backbone regularization, and custom restraints. QRNAS is capable of handling RNA, DNA, chimeras and hybrids thereof, and enables modeling of nucleic acids containing modified residues.
The POLYVIEW-2D protein structure visualization server can be used to generate amino acid sequence annotations, such as secondary structure, relative solvent accessibility, evolutionary conservation, and physico-chemical property profiles. It can also be used to identify residues involved in protein-protein interactions and highlight other important sites and motifs.
POLYVIEW-3D is a web-based tool for macromolecular structure visualization and analysis. In particular, it provides a wide array of options for automated structural and functional analysis of proteins and their complexes. This tutorial aims to describe and illustrate the available rendering options and annotation capabilities of POLYVIEW-3D.
POLYVIEW-MM (Molecular Motion) enables animation of trajectories generated by Molecular Dynamics and related simulation techniques, as well as visualization of alternative conformers, e.g., obtained as a result of protein structure prediction methods or small molecule docking, using an intuitive web-based interface.