Tracembler takes one or multiple DNA or protein sequence(s) as input to the NCBI Trace Archive BLAST engine to identify matching sequence reads from a species of interest. The BLAST searches are carried out recursively such that BLAST matching sequences identified in previous rounds of searches are used as new queries in subsequent rounds of BLAST searches. Tracembler streamlines the process of recursive database searches, sequence assembly, and gene identification in resulting contigs in attempts to identify homologous loci of genes of interest in species with emerging whole genome shotgun reads.
PacBio sequencers produced two types of characteristic reads: CCS (short and low error rate) and CLR (long and high error rate), both of which could be useful for de novo assembly of genomes. PBSIM simulates those PacBio reads by using either a model-based or sampling-based simulation.
hapAssembly beats the previous best for the important Haplotype Assembly Problem. It is an approach to finding optimal solutions for the haplotype assembly problem under the minimum-error-correction (MEC) model.
FastMLST is a high speed standalone script wrote in Python3, which takes assemblies in FASTA format (gzipped is also allowed) and determines its ST according to MLST schemes defined in PubMLST.
Ghosh, Priyanka, Sriram Krishnamoorthy, and Ananth Kalyanaraman.
“PaKman: A Scalable Algorithm for Generating Genomic Contigs on Distributed Memory Machines.”
IEEE Transactions on Parallel and Distributed Systems (TPDS) vol. 32, no. 5, pp. 1191-1209, 2021. DOI: 10.1109/TPDS.2020.3043241.
Trinity represents a novel method for the efficient and robust de novo reconstruction of transcriptomes from RNA-Seq data. Trinity combines three independent software modules: Inchworm, Chrysalis, and Butterfly, applied sequentially to process large volumes of RNA-Seq reads. Trinity partitions the sequence data into many individual de Bruijn graphs, each representing the transcriptional complexity at at a given gene or locus, and then processes each graph independently to extract full-length splicing isoforms and to tease apart transcripts derived from paralogous genes.
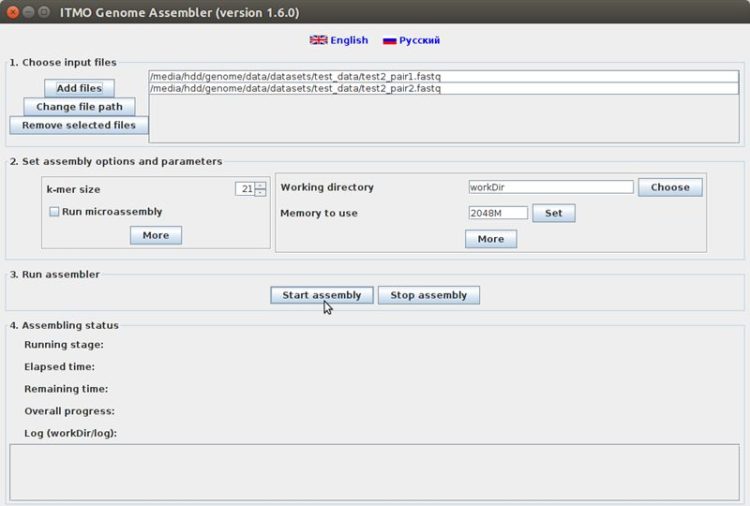
ITMO Genome Assembler is a tool for de novo assembly of small and middle-sized genomes from reads, produced by Illumina/Solexa, Ion Torrent or Sanger sequencers.