TopPIC is a software tool for identification and characterization of proteoforms at the whole proteome level by top-down tandem mass spectra using database search.
Protein identification using top-down spectra.
Liu X, Sirotkin Y, Shen Y, Anderson G, Tsai YS, Ting YS, Goodlett DR, Smith RD, Bafna V, Pevzner PA.
Mol Cell Proteomics. 2012 Jun;11(6):M111.008524. doi: 10.1074/mcp.M111.008524.
myProMS is a comprehensive bioinformatics environment (database and web server) for management of Mass Spectrometry (MS) protein identification data generated by database-search engines such as Mascot or Sequest. Multiple functionalities are available to mine, validate and interpret the data from both MS and biological point of views. In particular, biological interpretation of the results is facilitated through the use of sophisticated data comparison modules, annotation enrichments and links to external resources. myProMS was designed to optimize data access and sharing during collaboration between users with complementary expertises; typically MS experts and biologists.
ProteoSAFe is a Proteomics Environment which is Scalable in utilizing distributed computing, Accessible via reconfigurable, easy-to-learn user interfaces, and Flexible in tool chaining.
Inspect is a general purpose database search algorithm, with an emphasis on efficiently and confidently identifying modified peptides. It includes special scoring models for phosphorylation which allow for increased accuracy. In addition, Inspect implements the MS-Alignment algorithm for discovery of unanticipated modifications in blind mode.
pFind Studio is a computational solution for mass spectrometry-based proteomics. pFind Studio includes pFind, pBuild, pLabel, pXtract, pParse and pScan
pXtract creates .DTA .MGF and .MS2 input files directly from Thermo Scientific .raw LC-MS/MS data files.
pParse is a software dedicated to recalibrate the monoisotopic of precursors in MS/MS spectra datasets assigned by mass spectrometry
pFind is a search engine for peptide and protein identification via tandem mass spectrometry.
pNovo+ is a de novo peptide sequencing algorithm using complementary HCD and ETD tandem mass spectra.
pBuild is a tool that can compare several search engines’ results and combine them together. The latest version, pBuild v2.0, can process the search results of pFind, SEQUEST and Mascot.
pQuant is the software for quantitative proteomics, evaluates the accuracy of caluculated peptide and protein ratios.
pLabel is a spectra labeling tool that can visualize the global- and local-view peptide-spectrum matches, given the results of pFind or any other search engines. pLabel can label both CID and ETD spectra, and implement the manual de novo sequencing.
pLink is a software dedicated for the analysis of chemically cross-linked proteins or protein complexes using mass spectrometry.
pScan is a flexible tool that helps biologists to preprocess protein sequence databases in proteomics research.
pCluster is a software tool aiming at detecting protein modifications independent of sequence databases by tandem mass spectral clustering.
pMatch is a spectral library search tool, which is deliberately designed for the open search mode.
::DEVELOPER
Bioinformatics group, Institute of Computing Technology, Chinese Academy of Sciences
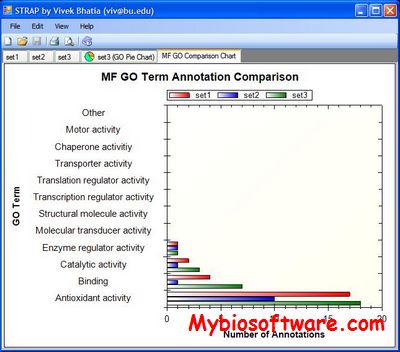
STRAP, the Software Tool for Rapid Annotation of Proteins, saves you time by automatically annotating a protein list with information that helps you meaningfully interpret your mass spectrometry data.
GoMiner organizes and allows the visualization of large sets of genes based on Gene Ontology classifications.GoMiner is a tool for biological interpretation of ‘omic’ data – including data from gene expression microarrays. Omic experiments often generate lists of dozens or hundreds of genes that differ in expression between samples, raising the question
DiagnoProt is a tool that finds discriminative mass spectra among different biological conditions and ultimately performs spectral profiling classification of unknown conditions by comparing sets of tandem mass spectra (MS/MS).
Silva ARF, Lima DB, Leyva A, Duran R, Batthyany C, Aquino PF, Leal JC, Rodriguez JE, Domont GB, Santos MDM, Chamot-Rooke J, Barbosa VC, Carvalho PC. DiagnoProt: a tool for discovery of new molecules by mass spectrometry.
Bioinformatics. 2017 Jun 15;33(12):1883-1885. doi: 10.1093/bioinformatics/btx093. PMID: 28186229.