MetaPepSeq is metaserver for protein identification in Mass Spectrometry Experiments. It helps joining the power of Mascot and de novo sequencing algorithms by merging together into one tool.
ALL-P is a statistical framework based on a hierarchical modeling that takes into account shared peptide information for estimating protein abundances. ALL-P performs a simultaneous analysis of all the quantified peptides, handling the biological and technical errors as well as the peptide effect.
::DEVELOPER
pappso (Plate-forme d’analyses protéomiques de Paris Sud-Ouest)
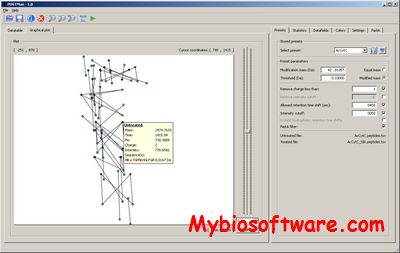
POSTMan is a software application developed at PROBE in cooperation with Stix AS, designed to aid the researcher in identifying post-translationally modified peptides present in a given sample. POSTMan aligns LC-MS runs (MS1 data) and isolates pairs of peptides which differ by a user defined specific mass difference (Post-translationally modified peptides). Candidate peptides can then be targeted for fragmentation in a second round of analysis allowing the identification of low abundant post-translationally modified peptides.
ProClassify is a tool for proteomic data classification. It was intended originally for high-resolution mass-spectrometry data classification, but it can be of use for datasets of a completely different nature as well.
atBioNet is a free, user friendly, web-based network analysis tool for analyzing, visualizing, and interpreting genomics or proteomics data. The user supplies atBioNet with a list of proteins or genes, and atBioNet then creates an interactive graphical network model that can identify key functional modules. Pathway information from the Kyoto Encyclopedia of Genes and Genomes (KEGG) is directly integrated within atBioNet for enrichment analysis and assessment of the biological meaning of modules.
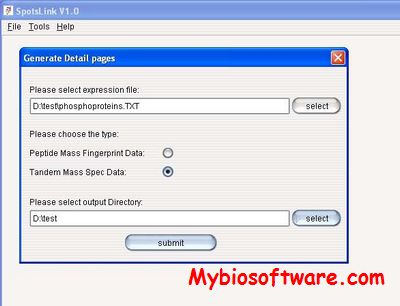
SpotLink is the tool for organizing proteomics raw data. The data includes 2D gel images, Mass Spectrometry (MS and MS/MS), protein expression profiles and etc.. The data is collected after a series of proteomics experiment and categorized by individual spot on gel.
Kim-Anh Do, Peter Mueller, Raj Bandyopadhya, Keith Baggerly (2008).
A Bayesian mixture model for protein biomarker discovery
Bayesian Modeling in Bioinformatics, Dey DK, Ghosh D, Mallick B (Eds), Chapman & Hall / CRC press
The Open2Dprot project is a community effort to create an open source n-dimensional (n-D) protein expression data pipeline-analysis system. It is downloadable and could be used for exploratory data analysis of protein expression data across sets of n-dimensional (n-D) data from research experiments. In the initial phase, as early beta-level versions of the pipeline modules are created, they are made available for download and may be used for performing parts of the analysis. Interchangeable subproject modules are being developed for 2-dimensional data including 2D-PAGE (polyacrylamide gel electrophoresis). Initial support for 2D LC-MS, protein arrays and other data will also be provided. In the second phase, it will be expanded to handle data from other n-dimensional protein separation methods as well as more extensive analyses on these data.