DiffSplice is a novel tool for discovering and quantitating alternative splicing variants present in an RNA-seq dataset, without relying on annotated transcriptome or pre-determined splice pattern. For two groups of samples, DiffSplice further utilizes a non-parametric permutation test to identify significant differences in expression at both gene level and transcription level. DiffSplice takes as input the SAM files that supply the alignment of the RNA-seq reads on the reference genome, obtained from an RNA-seq aligner like MapSplice. The results of DiffSplice are summarized as a decomposition of the genome and can be visualized using the UCSC genome browser.
Myrna is a cloud computing tool for calculating differential gene expression in large RNA-seq datasets. Myrna uses Bowtie for short read alignment and R/Bioconductor for interval calculations, normalization, and statistical testing. These tools are combined in an automatic, parallel pipeline that runs in the cloud (Elastic MapReduce in this case) on a local Hadoop cluster, or on a single computer, exploiting multiple computers and CPUs wherever possible.
Frida (FRamework for Image Dataset Analysis) is image analysis software. Frida was developed by the Johns Hopkins University Tissue Microarray Core Facility. Frida is open source and written in 100% Java. Frida makes use of functionality from the NIH’s ImageJ application.
Cornish T, Morgan J, Gurel B, and De Marzo AM.
FrIDA: An open source framework for image dataset analysis.
Arch. Pathol. Lab. Med. 132:856 (2008). Originally presented at Advancing Practice, Instruction and Innovation Through Informatics: Pittsburgh, PA, 2007.
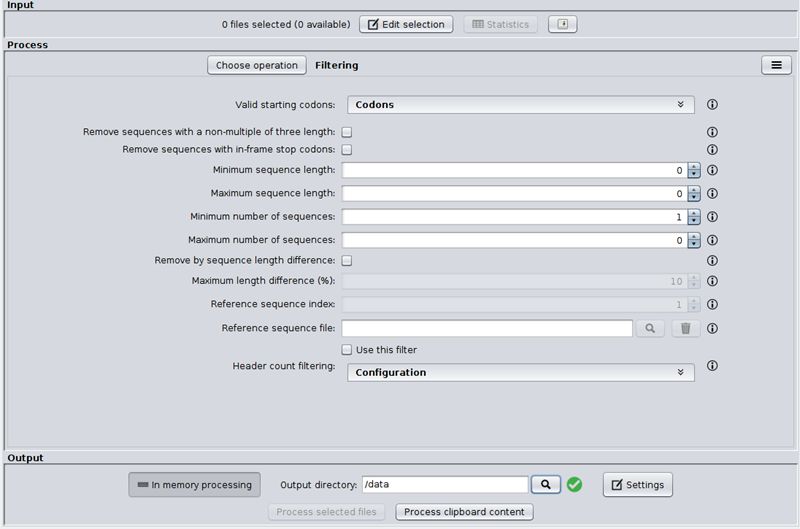
Lopez-Fernandez H, Duque P, Vazquez N, Fdez-Riverola F, Reboiro-Jato M, Vieira CP, Vieira J. SEDA: a Desktop Tool Suite for FASTA Files Processing.
IEEE/ACM Trans Comput Biol Bioinform. 2020 Nov 25;PP. doi: 10.1109/TCBB.2020.3040383. Epub ahead of print. PMID: 33237866.
HAPGEN simulates case control datasets at SNP markers. The new version can now simulate multiple disease SNPs on a single chromosome, on the assumption that each disease SNP acts independently and are in Hardy-Weinberg equilibrium. We also supply a R package that can simulate interaction between the disease SNPs.
ccPDB is designed to provide service to scientific community working in the field of function or structure annoation of proteins. This database of datasets is based on Protein Data Bank (PDB), where all datasets were derived from PDB. ccPDB have four modules; i) compilation of datasets, ii) creation of datasets, iii) web services and iv) Important links.