Baescs 2.0
:: DESCRIPTION
Baescs is a Bayesian approach to estimate calibration curves and unknown concentrations in immunoassays.
::DEVELOPER
The Center for Computational Immunology
:: SCREENSHOTS
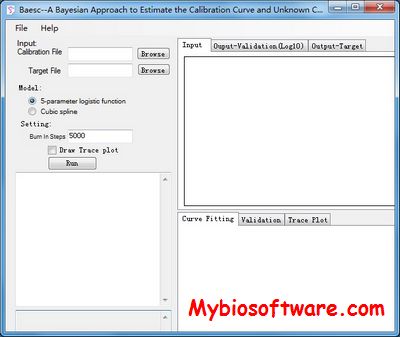
::REQUIREMENTS
- Linux/Windows
:: DOWNLOAD

:: MORE INFORMATION
Citation
Feng Feng; Ana Paula Sales; Thomas B. Kepler :
A Bayesian approach for estimating calibration curves and unknown concentrations in immunoassays.
Bioinformatics (2011) 27 (5): 707-712.