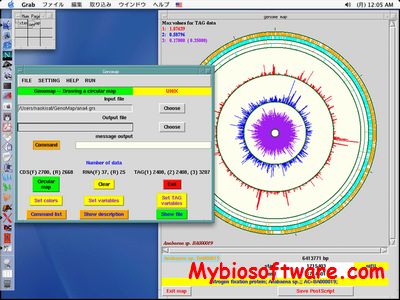
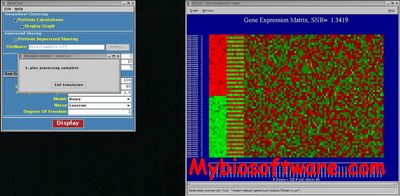
TightClust applies K-means clustering as an intermediate clustering engine. Early truncation of a hierarchical clustering tree is used to overcome the local minimum problem in K-means clustering. The tightest and most stable clusters are identified in a sequential manner through an analysis of the tendency of genes to be grouped together under repeated resampling.
ErmineJ performs analyses of gene sets in expression microarray data. A typical goal is to determine whether particular biological pathways are “doing something interesting” in the data. The software is designed to be used by biologists with little or no informatics background.
LIMMA is a software package for the analysis of gene expression microarray data, especially the use of linear models for analysing designed experiments and the assessment of differential expression. The package includes pre-processing capabilities for two-colour spotted arrays. The differential expression methods apply to all array platforms and treat Affymetrix, single channel and two channel experiments in a unified way.
Smyth, G. K. (2005). Limma: linear models for microarray data.
Bioinformatics and Computational Biology Solutions using R and Bioconductor, R. Gentleman, V. Carey, S. Dudoit, R. Irizarry, W. Huber (eds.),
Springer, New York, pages 397-420
GeneClust is a piece of computer software which can be used as a tool for exploratory analysis of gene expression microarray data. The development of GeneClust was motivated by surging interest to search for interpretable biological structure in gene expression microarray data.
CpGassoc – An R Function for Analysis of DNA Methylation Microarray Data The analysis of DNA methylation data has recently garnered attention among researchers from a variety of backgrounds, due to the availability of high-throughput methylation microarrays. The number of CpG sites that can be analyzed is growing rapidly – for example, the latest Illumina Infinium BeadChip interrogates ~450,000 CpG sites. With the growing interest in DNA methylation and the growing volume of data analyzed, there is a need for software to perform these types of analyses.
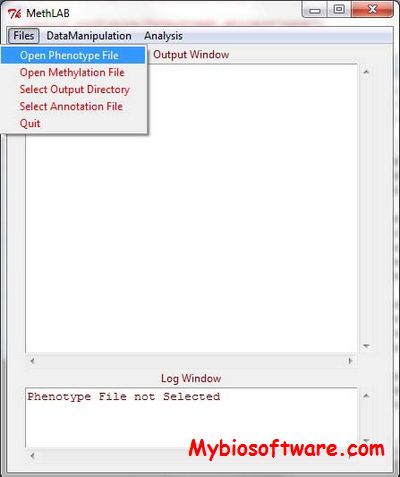
MethLAB provides a graphical user interface (GUI) to facilitate analysis of DNA methylation microarray data, allowing users with no experience using statistical software to implement flexible and powerful analyses of array-based DNA methylation data.
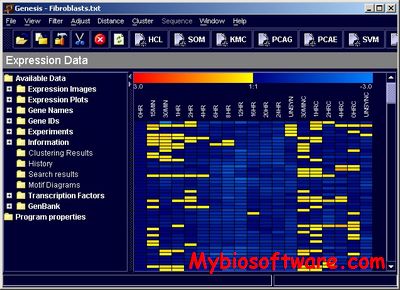
Genesis integrates various tools for microarray data analysis such as filters, normalization and visualization tools, distance measures as well as common clustering algorithms including hierarchical clustering, self-organizing maps, k-means, principal component analysis, and support vector machines.
Genesis Server is an application server for computation of Hierarchical Clustering, Self Organizing Maps (SOM), k-means Clustering and Support Vector Machines (SVM).
Backes C, Keller A, Kuentzer J, Kneissl B, Comtesse N, Elnakady YA, Müller R, Meese E, Lenhof HP. GeneTrail–advanced gene set enrichment analysis.
Nucleic Acids Res. 2007 Jul;35(Web Server issue):W186-92.