HapCART is a convenient analysis tool for detecting disease-related haplotype-haplotype interactions. HapCART overcome high-dimensional issues which combine the advantages of data mining with the concept of haplotypes and consider haplotype uncertainty.
::DEVELOPER
Cathy S.J. Fann lab,Institute of Biomedical Informatics, National Yang-Ming University, Taipei
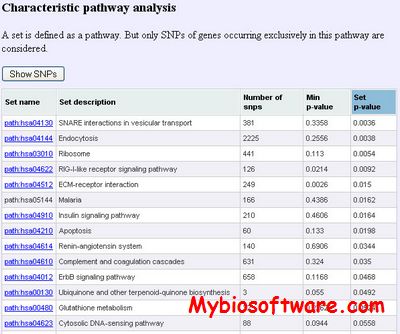
GWAS Pathway Identifier combines GWAS(Genome-Wide Accociation Studies) and pathway data as well as known and predicted protein-interaction data to identify disease specific SNP sets.
VAAST (the Variant Annotation, Analysis and Search Tool) is a probabilistic search tool for identifying damaged genes and their disease-causing variants in personal genome sequences. VAAST builds upon existing amino acid substitution (AAS) and aggregative approaches to variant prioritization, combining elements of both into a single unified likelihood-framework that allows users to identify damaged genes and deleterious variants with greater accuracy, and in an easy-to-use fashion. VAAST can score both coding and non-coding variants, evaluating the cumulative impact of both types of variants simultaneously. VAAST can identify rare variants causing rare genetic diseases, and it can also use both rare and common variants to identify genes responsible for common diseases. VAAST thus has a much greater scope of use than any existing methodology.
DigSee is a text mining search engine to provide evidence sentences describing that “genes” are involved in the development of “disease” through “biological events”. Biological events such as gene expression, regulation, phosphorylation, localization, and protein catabolism play important roles in the development of diseases. Understanding the association between diseases and genes can be enhanced with the identification of involved biological events in this association. With input of (disease, genes, events), users can obtain Medline abstracts with highlighted evidence sentences.
DAPPLE looks for significant physical connectivity among proteins encoded for by genes in loci associated to disease according to protein-protein interactions reported in the literature. The hypothesis behind DAPPLE is that causal genetic variation affects a limited set of underlying mechanisms that are detectable by protein-protein interactions.
gTDT implemented gene-based or group-wise TDT for rare variant aggregation analysis. Currently gTDT implemented haplotype-based tests for 6 models, M1-M6. It takes as input a ped file and a dat file that specify the relationships, and a VCF file that stores genotype data.
LRASSOC suite deals with the situation where we have a case-control sample of affected and unaffected individuals with their marker genotypes for 2 biallelic marker loci. These 2 marker loci may be in linkage disequilibrium with 1 or 2 biallelic disease susceptibility loci and therefore affect disease risk through association or may themselves be disease susceptibility loci. We are interested in modelling the effects of the genotype on the probability of disease risk in order to draw conclusions regarding the nature of the joint effect of the loci. Among the issues we may wish to investigate are whether either of the 2 loci actually has an effect on disease risk, the strength and statistical significance of any effect, the nature of such an effect e.g is the effect additive on some scale or do the alleles at the same loci interact in a dominance effect. We also want to compare single and joint locus models to investigate how the strength and significance of the effect of each locus is affected by the presence or absence of the other in a model and, a related point, whether the additive and dominance effects of two loci are independent or whether they interact (often called epistasis in this context).