CrypticIBDcheck 0.3-3
:: DESCRIPTION
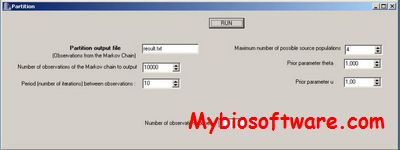
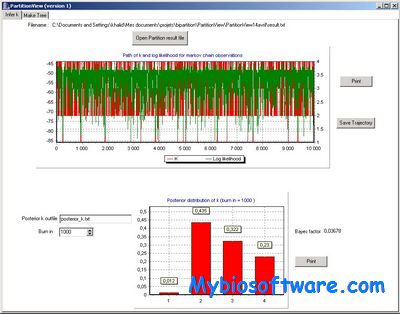
CrypticIBDcheck can be used to identify pairs of closely-related subjects based on genetic marker data from single-nucleotide polymorphisms (SNPs). The package is able to accommodate SNPs in linkage disequibrium (LD), without the need to thin the markers so that they are approximately independent in the population. Sample pairs are identified as related by superposing their estimated identity-by-descent (IBD) coefficients on plots of IBD coefficients for pairs of simulated subjects from one of several common close relationships.
::DEVELOPER
: SCREENSHOTS
N/A
:: REQUIREMENTS
- Linux / Windows / MacOsX
- R Package

:: MORE INFORMATION
Citation
Source Code Biol Med. 2013 Feb 6;8(1):5. doi: 10.1186/1751-0473-8-5.
CrypticIBDcheck: an R package for checking cryptic relatedness in nominally unrelated individuals.
Nembot-Simo A, Graham J, McNeney B.