PTest (Partition test) is a software for case-control genetic association studies in human gene mapping. The PTest is based on single-SNP p-values resulting from an association test, for example, the chi-square test comparing genotype or allele frequencies in cases and controls.
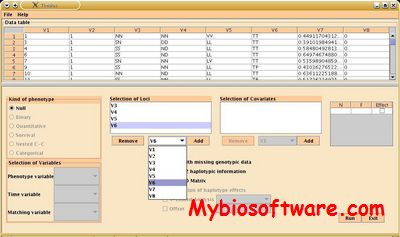
The objectif of the THESIAS program is to performed haplotype-based association analysis in unrelated individuals. This program is based on the maximum likelihood model.THESIAS allows one to simultaneous estimate haplotype frequencies and their associated effects on the phenotype of interest. In this new THESIAS release, quantitative, qualitative (logistic and matched-pair analysis), categorical and survival outcomes can be studied. X-linked haplotype analysis is also feasible.
::DEVELOPER
Tregouet David <tregouet@chups.jussieu.fr>
Garelle Valérie (garelle@chups.jussieu.fr)
Expression levels of mRNAs are among other factors regulated by microRNAs. A particular microRNA can bind specifically to several target mRNAs and lead to their degradation. Expression levels of both, mRNAs and microRNAs, can be obtained by microarray experiments. In order to increase the power of detecting microRNAs that are differentially expressed between two different groups of samples, “miRtest” incorporates expression levels of their related target gene sets.
Broad-Enrich tests sets of broad genomic regions (e.g., from ChIP-seq data for histone modifications or copy number variations) for enriched biological pathways, Gene Ontology terms, or other gene sets.
Tarsier is a software pipeline for testing RNA gene prediction programs with genomic alignments. It automatically gathers RNA data from Rfam and genomic alignments from both UCSC and Ensembl, and executes two popular prediction programs, EvoFold and RNAz.
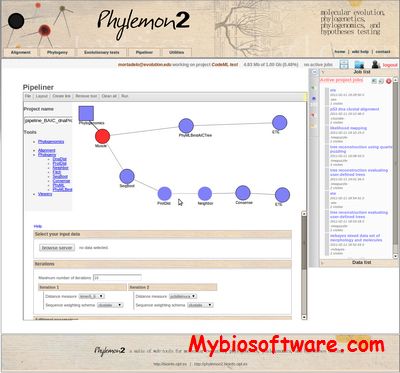
Phylemon is the suite of web-tools for molecular evolution, phylogenetics and phylogenomics. It is conceived as a natural response to the increasing demand of data analysis of experimental scientists seeking to add molecular evolution and phylogenetic insight into their research.
IBMT is a Bayesian hierarchical normal model to define a novel Intensity-Based Moderated T-statistic.The method is completely data-dependent using empirical Bayes philosophy to estimate hyperparameters, and thus does not require specification of any free parameters. IBMT has the strength of balancing two important factors in the analysis of microarray data: the degree of independence of variances relative to the degree of identity (i.e. t-tests vs. equal variance assumption), and the relationship between variance and signal intensity. When this variance-intensity relationship is weak or does not exist, IBMT reduces to a previously described moderated t-statistic. Furthermore, our method may be directly applied to any array platform and experimental design. Together, these properties show IBMT to be a valuable option in the analysis of virtually any microarray experiment.
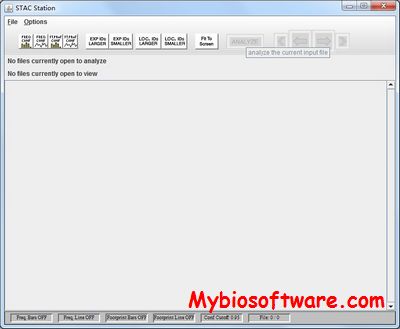
STAC (Significance Testing for Aberrant Copy-Number) is a method for testing the significance of DNA copy number aberrations across multiple array-CGH experiments. It utilizes two complementary statistics in combination with a novel search strategy. The significance of both statistics is assessed, and P-values are assigned to each location on the genome by using a multiple testing corrected permutation approach. STAC identifies genomic alterations known to be of clinical and biological significance and provides statistical support for 85% of previously reported regions. Moreover, STAC identifies numerous additional regions of significant gain/loss in these data that warrant further investigation. The P-values provided by STAC can be used to prioritize regions for follow-up study in an unbiased fashion.