The Tumorscape portal facilitates the use and understanding of high resolution copy number data amassed from multiple cancer types. It supports gene-level analysis, analysis by cancer type, and the downloading/browsing of data.
BASTA contains an implementation of a Bayesian model-based method for unsupervised classification of tissue samples based on DNA copy number amplifications.
MPCBS (Multi-Platform Circular Binary Segmentation) is a software which pools statistical evidence across platforms during segmentation, and does not require pre-standardization of different data sources. It involves a weighted sum of t-statistics, which arises naturally from the generalized log-likelihood ratio of a multi-platform model.
PSCN segments the genome of a sample into homogeneous parts and gives an estimation of parent-specific DNA copy number using high-density SNP array data (logR and B-allele frequency). This package can be applied on platforms having both SNP probes and copy number probes.
CNAnova is a stand-alone software package for identifying recurrent regions of copy number aberrations (CNAs) using SNP microarray data. It runs from the command line on the Linux platforms and is composed of several modules written in the R programming language.
STAC (Significance Testing for Aberrant Copy-Number) is a method for testing the significance of DNA copy number aberrations across multiple array-CGH experiments. It utilizes two complementary statistics in combination with a novel search strategy. The significance of both statistics is assessed, and P-values are assigned to each location on the genome by using a multiple testing corrected permutation approach. STAC identifies genomic alterations known to be of clinical and biological significance and provides statistical support for 85% of previously reported regions. Moreover, STAC identifies numerous additional regions of significant gain/loss in these data that warrant further investigation. The P-values provided by STAC can be used to prioritize regions for follow-up study in an unbiased fashion.
GMM (Gaussian Mixture Model) detects copy number variation from the distribution of copy number ratios. From the data, it will fit one component for each of the following copy number states: deletion, copy-neutral, 1 and 2 additional copy; with a constraint on the difference between the mixture means. Then for a given individual, it will determine the probabilities for each copy number state and compute the expected copy number (dosage).
CNSuite (Copy Number Suite) is a caBIGTM (cancer Biomedical Informatics Grid) analytical tool for gene copy number change analysis. CNSuite consists of a Fused Margin Regression (FMR) method for detecting copy number changes in a single signal profile and consensus copy number changes in population data, and two feature indexing methods for analyzing chromosomal instabilities (CIN). CNSuite is applicable to analyzing germline copy number variations (CNV) in the study of population genetics and somatic copy number alterations (CNA) in tumor genomics.
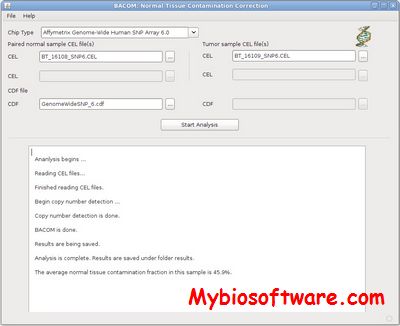
BACOM (Bayesian Analysis of COpy number Mixtures) is a statistically-principled in silico approach to accurately estimate genomic deletions and normal tissue contamination, and accordingly recover the true copy number profile in cancer cells. We have developed a cross-platform and open source Java application that implements the whole pipeline of copy number analysis of heterogeneous cancer tissues and other relevant processing steps. We also provide an R interface, bacomR, for running BACOM within the R environment, through which users can smoothly incorporate BACOM into their specific analyses.