SVAMP 2.10
:: DESCRIPTION
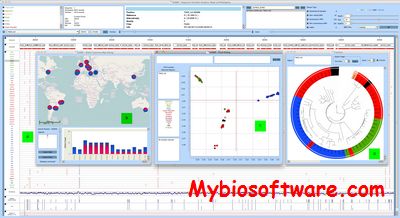
SVAMP is standalone desktop application to visualise variants (SNPs and indels) and peform realtime analysis on selected regions of a genome on specific samples. SVAMP has some interesting features like phylogeography,allele frequency map and principal coordinate analysis.
::DEVELOPER
Computational Bioscience Research Center, King Abdullah University of Science and Technology
:: SCREENSHOTS
:: REQUIREMENTS
- Linux / MacOsX
- Perl
:: DOWNLOAD

:: MORE INFORMATION
Citation
Bioinformatics. 2014 Apr 23. [Epub ahead of print]
SVAMP: sequence variation analysis, maps and phylogeny.
Naeem R1, Hidayah L, Preston MD, Clark TG, Pain A.