REPRO (Protein Repeats Analysis) is able to recognise distant repeats in a single query sequence. The technique relies on a variation of the Smith-Waterman local alignment strategy to find non-overlapping top-scoring local alignments, followed by a graph-based iterative clustering procedure to delineate the repeat set(s) based on consistency of the pairwise top-alignments.
primeGEM is a program that performs probabilistic orthology analysis (Arvestad et al. 2003, 2004, submitted, Sennblad and Lagergren, 2009). It computes for a pair of genes the probability that they are orthologous (note that this correpsponds to the probability that their least common ancestor is a speciation). It is based on the gene evolution model which models gene duplication and loss using a generalized birth-death model with threee parameters. The parameters can be integrated over using MCMC or (soon) maximized over using ML.
CoCAS is a standalone Chromatin immunoprecipitation microarray (ChIP-on-chip) analysis application. It has been designed to be used primarily on Agilent microarrays scanned with an Agilent scanner.
CoCAS can readily be used on the latest generation of Agilent high-density arrays. Unlike other tools available, Cocas allows dye-swap, replicate correlation and connects easily with other software (genome browsers and peak detection). The peak detection methods implemented in CoCAS can be used for any set of data including ChIP-Seq data.
Picky is an oligo design program that alleviates long and monotonous calculations by identifying probes that are very unique and specific to sequence regions. These calculations are based on parameters inputted by the user including optimal probe length, ideal percentage of guanine and cytosine content, salt concentration and the maximum length to which a target sequence matches any non-target sequence.
PICKY allows the rapid and efficient determination of gene-specific oligos based on given gene sets, and can be used for large, complex genomes such as human, mouse, or maize. PICKY can also be used to analyze existing microarray probes and evaluate their design quality. Because PICKY uses rigorous whole genome-based thermodynamic screening to identify potential hydrogen binding sites of each probe, it can also be utilized to design short interference RNA (siRNA) for gene knockout applications and to discover possible targets of regulatory short RNAs (e.g., microRNAs or sRNAs). Picky has exceptional computational capability and is also user friendly.
If you find Picky useful to your research, please cite the Picky papers below that are most relevant to your work. If you have no preference, we recommend you cite the newest Shared Probe Design paper.
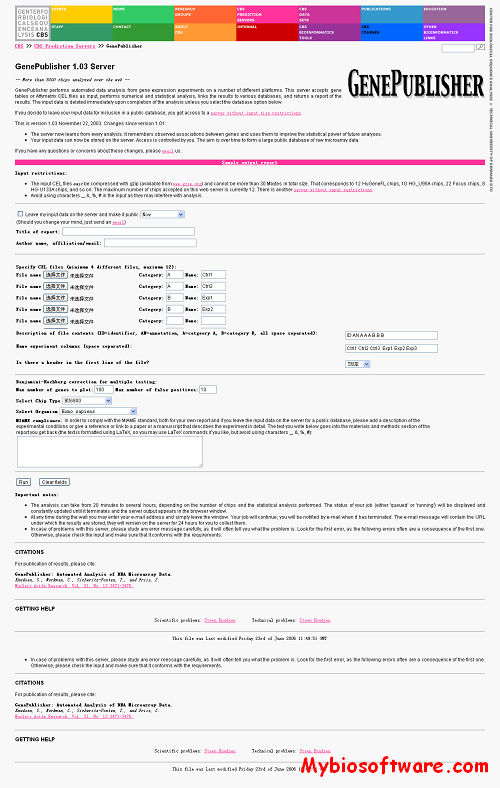
GenePublisher performs automated data analysis from gene expression experiments on a number of different platforms. This server accepts gene tables or Affymetrix CEL files as input, performs numerical and statistical analysis, links the results to various databases, and returns a report of the results. The input data is deleted immediately upon completion of the analysis unless you select the database option below.
::DEVELOPER
CBS (Center for Biological Sequence Analysis) at Technical University of Denmark
:: SCREENSHOTS
:: REQUIREMENTS
web browser
:: MORE INFORMATION
Input restrictions:
The input CEL files must be compressed with gzip (available from www.gzip.org) and cannot be more than 30 Mbytes in total size. That corresponds to 12 HuGeneFL chips, 10 HG_U95A chips, 22 Focus chips, 8 HG-U133A chips, and so on. The maximum number of chips accepted on this web server is currently 12. There is another server without input restrictions.
Avoid using characters _, &, %, # in the input as they may interfere with analysis.
AMIADA is an integrated computer program for organizing, exploring, visualizing, and analyzing microarray data. It features an EXCEL-like user interface and performs data transformation, principal component analysis, a variety of cluster analysis and extensive graphic functions for visualizing expression profiles.
SilVA (Latin for “forest”) is a tool for the automated harmfulness prediction of synonymous (silent) mutations within the human genome. SilVA bases its predictions on a number of features, including conservation, codon usage, splice sites, splicing enhancers and suppressors, and mRNA folding free energy. Given variants in a VCF file, SilVA will rank the rare synonymous variants according to their predicted harmfulness.