xPyder 201208
:: DESCRIPTION
xPyder is a PyMOL plugin to analyze and visualize on the 3D structure dynamical cross-correlation matrices (DCCM), linear mutual information (LMI), communication propensities (CP), intra- and inter-molecular interactions (e.g. PSN), and more, to produce highly customizable publication-quality images.
::DEVELOPER
:: SCREENSHOTS
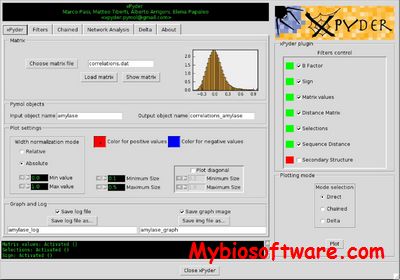
:: REQUIREMENTS
- Linux /Windows/ MacOsX
- Python
- PyMol
:: DOWNLOAD

:: MORE INFORMATION
Citation
xPyder: a PyMOL plugin to analyze coupled residues and their networks in protein structures.
Pasi M, Tiberti M, Arrigoni A, Papaleo E.
J Chem Inf Model. 2012 Jul 23;52(7):1865-74. doi: 10.1021/ci300213c