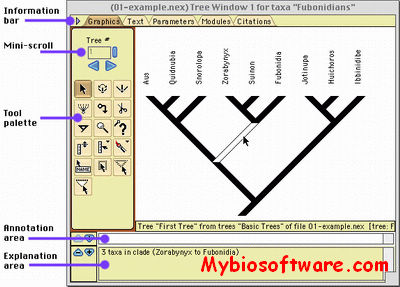
Topali (tree TOPology-related analysis of ALignments Interface) is a software for statistical and evolutionary analysis of multiple sequence alignments.The extended TOPALi v2 provides phylogenetic model selection, Bayesian analysis (BA) and Maximum Likelihood (ML) phylogenetic tree estimation, detection of sites under positive selection, and recombination breakpoint location analysis.
Mesquite is software for evolutionary biology, designed to help biologists analyze comparative data about organisms. Its emphasis is on phylogenetic analysis, but some of its modules concern population genetics, while others do non-phylogenetic multivariate analysis. Because it is modular, the analyses available depend on the modules installed.
ConPlex is a web application for evolutionary analysis of protein complex structures. With given protein complex structures, ConPlex automatically identifies protein interfaces and carries out evolutionary conservation analyses for the identified regions.
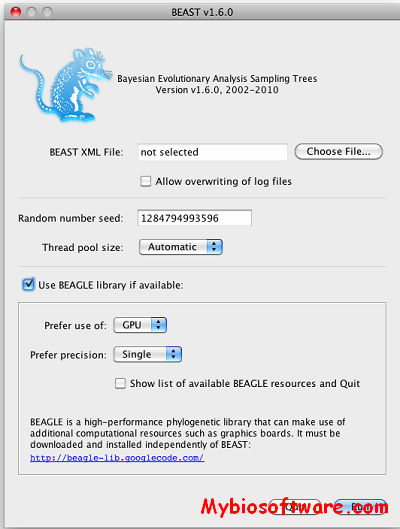
BEAST (Bayesian Evolutionary Analysis Samling Trees) is a cross-platform program for Bayesian MCMC analysis of molecular sequences. It is entirely orientated towards rooted, time-measured phylogenies inferred using strict or relaxed molecular clock models. It can be used as a method of reconstructing phylogenies but is also a framework for testing evolutionary hypotheses without conditioning on a single tree topology. BEAST uses MCMC to average over tree space, so that each tree is weighted proportional to its posterior probability. We include a simple to use user-interface program for setting up standard analyses and a suit of programs for analysing the results.
BEAST 2 is an open source cross-platform program for Bayesian MCMC phylogenetic analysis of molecular sequences.
BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis.
Bouckaert R, Vaughan TG, Barido-Sottani J, Duchêne S, Fourment M, Gavryushkina A, Heled J, Jones G, Kühnert D, De Maio N, Matschiner M, Mendes FK, Müller NF, Ogilvie HA, du Plessis L, Popinga A, Rambaut A, Rasmussen D, Siveroni I, Suchard MA, Wu CH, Xie D, Zhang C, Stadler T, Drummond AJ.
PLoS Comput Biol. 2019 Apr 8;15(4):e1006650. doi: 10.1371/journal.pcbi.1006650.
Count is a software package for the evolutionary analysis of homolog family sizes (phylogenetic profiles), or other numerical census-type characters along a phylogeny. The principal data consist of the distribution of homolog family sizes across multiple genomes: for each (gene) family, that table gives the number of homologs identified in each genome.