BALM ( bi-asymmetric-Laplace model) was developed to analyze MBD-seq. We compared the resolution of BALM to that of several ChIP-seq analysis tools. The results demonstrate the program’s superior ability to distinguish methylation statuses of closely positioned CpG sites. This study demonstrates that MBD-seq combined with the new program is potentially a powerful tool to capture genome-wide DNA methylation profiles with high efficiency and resolution.
FlowAnd is designed to analyze and integrate largescale, multi-color flow cytometry data. It implements methods for data importing, various transformations, several clustering algorithms for automatic clustering, visualization tools as well as straightforward statistical testing.
Trans-ABySS is a software pipeline for analyzing ABySS-assembled contigs from shotgun transcriptome data. The pipeline accepts assemblies that were generated across a wide range of k values in order to address variable transcript expression levels. It first filters and merges the multi-k assemblies, generating a much smaller set of nonredundant contigs. It contains scripts that map assembled contigs to known transcripts, currently supporting Blat and Exonerate contig-to-genome aligners. It identifies novel splicing events like exon-skipping, novel exons, retained introns, novel introns, and alternative splicing. Its scripts can also estimate gene expression levels, identify candidate polyadenylation sites, and identify candidate gene-fusion events.
Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, Mungall K, Lee S, Okada HM, Qian JQ, Griffith M, Raymond A, Thiessen N, Cezard T, Butterfield YS, Newsome R, Chan SK, She R, Varhol R, Kamoh B, Prabhu AL, Tam A, Zhao Y, Moore RA, Hirst M, Marra MA, Jones SJ, Hoodless PA, Birol I. De novo assembly and analysis of RNA-seq data.
Nat Methods. 2010 Nov;7(11):909-12. Epub 2010 Oct 10.
STAMP (Statistical Analysis of Metagenomic Profiles) is a software package for analyzing metagenomic profiles (e.g., a taxonomic profile indicating the number of marker genes assigned to different taxonomic units or a functional profile indicating the number of sequences assigned to different biological subsystems or pathways) that promotes ‘best practices’ in choosing appropriate statistical techniques and reporting results. It encourages the use of effect sizes and confidence intervals in assessing biological importance. A user friendly, graphical interface permits easy exploration of statistical results and generation of publication quality plots for inferring the biological relevance of features in a metagenomic profile.
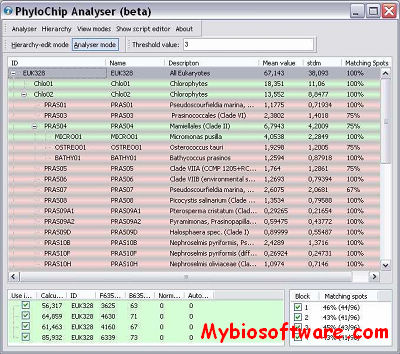
PhylochipAnalyzer is a Windows-program for the analysis of experiments with hierarchical probe-sets. It operates in two modes: first, the hierarchy of probes is defined interactively, second, the intensity data of a hybridized chip is loaded and analyzed according to the hierarchy. The program can export hierarchy trees to Newick-format and analyzed data to Excel. It contains a Delphi-script that makes it configurable with respect to different criteria for positive signals.
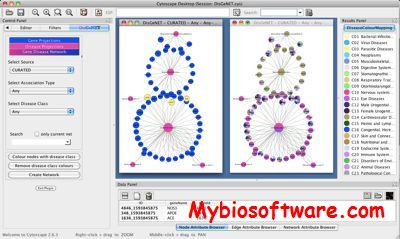
DisGeNET is a plugin for Cytoscape to query and analyze a network representation of human gene-disease databases. For this purpose, we have developed a new gene-disease database integrating data from several public sources. DisGeNET allows user-friendly access to our database, which includes queries restricted to (i) the original data source, (ii) the association type, (iii) the disorder class of interest and (iv) specific diseases, respectively genes. It represents gene-disease associations in terms of bipartite graphs and additionally provides gene centric and disease centric views of the data. It assists the user in the interpretation and exploration of human complex diseases with respect to their genetic origin by a variety of built-in functions. Moreover, DisGeNET permits multicoloring of nodes (genes/diseases) according to their disease classes for expedient visualization
DisGeNET2R contains a series of functions to retrieve and expand gene-disease, and variant-disease data, and to perform mappings of several biomedical vocabularies.