LIS alignment is a www interface for parsing BLAST output using the LIS (Longest Increasing Subsequence) algorithm. The softwar processes the BLAST result to find the longest consecutive list of HSPs.
SIB-BLAST (Simple Is Beautiful) is a novel algorithm developed to overcome the model corruption problem that occurs frequently in the later iterations of PSI-BLAST searches.The algorithm compares resultant hits from iteration two and the final iteration of a PSI-BLAST search, calculates the figure of merit for each “overlapped” hit and re-ranks the hits according to their figure of merit. The premise of the algorithm is based on the observation that the profile, namely, the position specific scoring matrix (PSSM), in the first two rounds of a PSI-BLAST search, is the least corrupted since it is comprised mostly of close homologs. These profiles are used to search for more distant homologs, which are used to generate subsequent PSSMs. As more distant homologs are incorporated into the PSSM, non-homologous sequences frequently get included also, thus leading to model corruption. Hence, “benchmarking” hits from later iteration against earlier round when the model is least corrupted should improve the accuracy of a PSI-BLAST search.
PTMClust is a software that can be applied to the output of blind PTM (Post-translational Modification) search methods to improve prediction quality, by suppressing noise in the data and clustering peptides with the same underlying modification to form PTM groups. We showed that our technique outperforms two standard clustering algorithms on a simulated dataset. Additionally, we showed that our algorithm significantly improves sensitivity and specificity when applied to the output of three different blind PTM search engines, SIMS, InsPecT and MODmap. Additionally, PTMClust markedly outforms another PTM refinement algorithm, PTMFinder. We demonstrate that our technique is able to reduce false PTM assignments, improve overall detection coverage and facilitate novel PTM discovery, including terminus modifications. We applied our technique to a large-scale yeast MS/MS proteome profiling dataset and found numerous known and novel PTMs. Accurately identifying modifications in protein sequences is a critical first step for PTM profiling, and thus our approach may benefit routine proteomic analysis.
The ISA (Iterative Signature Algorithm) was designed to reduce the complexity of very large sets of data by decomposing it into so-called “modules”. In the context of gene expression data these modules consist of subsets of genes that exhibit a coherent expression profile only over a subset of microarray experiments. Genes and arrays may be attributed to multiple modules and the level of required coherence can be varied resulting in different “resolutions” of the modular mapping. Since the ISA does not rely on the computation of correlation matrices (like many other tools), it is extremely fast even for very large datasets.
ABACAS (Algorithm Based Automatic Contiguation of Assembled Sequences) is intended to rapidly contiguate (align, order, orientate), visualize and design primers to close gaps on shotgun assembled contigs based on a reference sequence.
The Kinwalker algorithm performs cotranscriptional folding of RNAs, starting at a user a specified structure (default: open chain) and ending at the minimum free energy structure. Folding events are performed between transcription of additional bases and are regulated by barrier heights between the source and target structure.
J Mol Biol. 2008 May 23;379(1):160-73. Epub 2008 Mar 6. Folding kinetics of large RNAs.
Geis M, Flamm C, Wolfinger MT, Tanzer A, Hofacker IL, Middendorf M, Mandl C, Stadler PF, Thurner C.
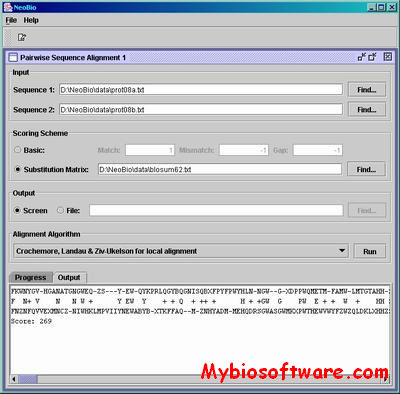
NeoBio is a Java class library of Computational Biology Algorithms. The current version consists mainly of pairwise sequence alignment algorithms such as the classical dynamic programming methods of Needleman-Wunsch and Smith-Waterman.