HiCNet
:: DESCRIPTION
HiCNet is an approach for establishing small-world networks for individual chromosomes using Hi-C data. It is a tool capable of converting Hi-C contacts to spatial distances. And it can also infer three-dimensional structures using spatial distances and multidimensional scaling methods.
::DEVELOPER
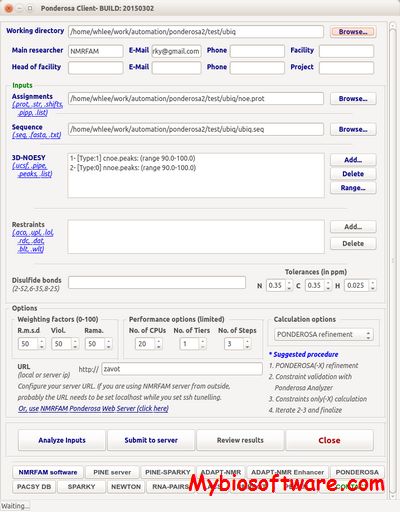
:: SCREENSHOTS
N/A
:: REQUIREMENTS
- Linux
- Perl
:: DOWNLOAD

:: MORE INFORMATION
Citation
Liu T, Wang Z.
Reconstructing high-resolution chromosome three-dimensional structures by Hi-C complex networks.
BMC Bioinformatics. 2018 Dec 28;19(Suppl 17):496. doi: 10.1186/s12859-018-2464-z. PMID: 30591009; PMCID: PMC6309071.