scHiCluster v0.1.1
:: DESCRIPTION
scHiCluster is a comprehensive python package for single-cell chromosome contact data analysis. It includes the identification of cell types (clusters), loop calling in cell types, and domain and compartment calling in single cells.
::DEVELOPER
:: SCREENSHOTS
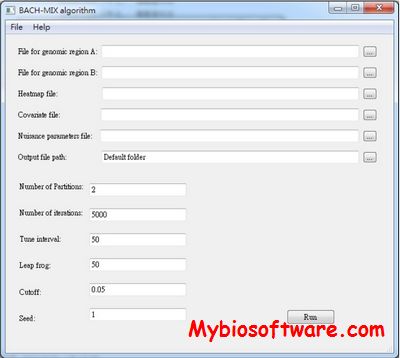
N/A
:: REQUIREMENTS
- Linux
- Python
:: DOWNLOAD

:: MORE INFORMATION
Citation:
Zhou J, Ma J, Chen Y, Cheng C, Bao B, Peng J, Sejnowski TJ, Dixon JR, Ecker JR.
Robust single-cell Hi-C clustering by convolution- and random-walk-based imputation.
Proc Natl Acad Sci U S A. 2019 Jul 9;116(28):14011-14018. doi: 10.1073/pnas.1901423116. Epub 2019 Jun 24. PMID: 31235599; PMCID: PMC6628819.