COMPONENT is a computer program for analysing evolutionary trees and is intended for use in studies of phylogeny, tree shape distribution, gene trees/species trees, host-parasite cospeciation, and biogeography.
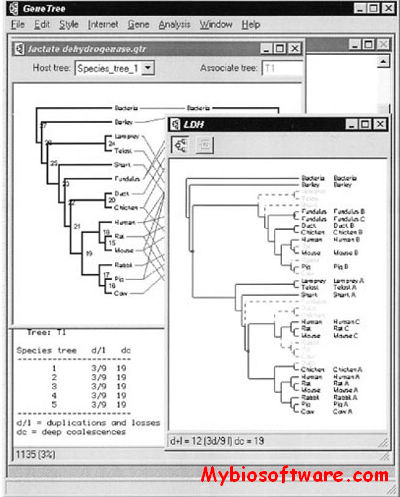
GeneTree is an experimental program for comparing gene and species trees. The program can compute the cost of embedding a gene tree within a species tree, visually display the location and number of gene duplications and losses, and search for optimal species trees.
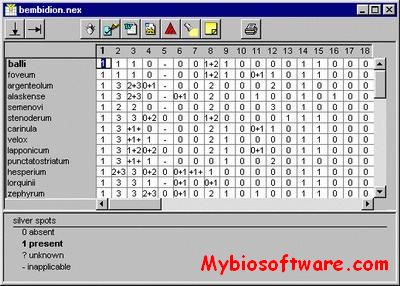
NDE (NEXUS Data Editor) is a program to create and edit NEXUS format data files on computers running Microsoft Windows 95/NT 4.0. The main motivation behind my writing the program was to provide an easy to use data editor similar to that provided in the Macintosh program MacClade (note that NDE has none of the data or tree analysis features of MacClade). The NEXUS format is becoming more widely used on PCs now that DOS and Windows versions of PAUP* are available for testing.
WINIPAUP (Windows Interface for PAUP) is an interface allowing the creation of NEXUS files including the data and the commands necessary to the realization of analyzes in PAUP software (D. SWOFFORD).WINIPAUP allows the importation of sequences (nucleotidic and proteic), discrete data, distances matrix, phylogenetic trees and NEXUS files.Moreover the software allows the creation of files which, carried out in PAUP, provide results files usable by MODELTEST software (D. POSADA).
Filo is designed to be used to simulate (molecular) sequence data used in phylogenetic analyses under very general conditions, including:
insertions and deletions; Insertions and deletions are propagated through the tree so the output file can be made to be the “true” alignment. This is very handy for checking out alignment methods, and is one of the main motivations for writing this program.
different phylogenetic histories (trees) along a sequence;
arbitrary rate matrices for each tree;
definition and re-use of node, branch, tree and global parameters;
output in various formats for subsequent analysis, e.g. in phylogenetic inference programs;
generation of trees under standard models such as Yule and All Trees Equally Likely.
This software is intended for use in generating simulated sequence data for phylogenetic analysis. It is released as is, and with no warranty of any kind. Use it at your own risk!
CoSpec (short for CoSpeciation) is a simple Macintosh Classic application that can be used to show the coevolutionary behaviours of two phylogenies, one dependent on the other.
Spectrum is a program designed to read in a data file comprising aligned character arrays (such as DNA sequence data) or distance data in the form of a matrix, and perform some simple spectral analysis procedures on that data.The input file format is NEXUS, as used in PAUP and MacClade.The program reads in phylogenetic 4-state or binary data, or distance data, and outputs the bipartition spectra corresponding to the data. It can also be used it to find the tree whose expected spectrum is closest to your observed spectrum (the “closest tree” and “Manhattan tree”– see the manual). Spectrum outputs spectra in Excel format as tab-delimited text files and trees as TreeView files.
Please register your copy of Spectrum! It costs nothing, and if you register I can keep you up to date with bugs, bug fixes, updates etc. E-mail me to register.
OrthoSelect is a easy-to-install and easy-to-use tool for finding ortholog groups in EST databases. It automatically searches assembled EST sequences against databases of ortholog groups (OG), assigns ESTs to these pre-defined OGs, translates the sequences into proteins, eliminates redundant sequences assigned to the same OG, creates multiple sequence alignments of identified ortholog sequences and others the possibility to further process this alignment in a last step. OrthoSelect performes better than the best-hit selection strategy and shows reliable results re-annotating database member sequences of OrthoMCL-DB and KOG. Since a correct orthology assignment is a important prerequisite for the construction of reliable data sets, OrthoSelect is capable of producing such data sets. This makes a OrthoSelect a valuable tool for researcher dealing with large EST libraries focussing on constructing data sets for phylogenetic reconstructions.
Treephyler is a tool for fast taxonomic profiling of metagenomes. It combines the predictive power of tree-based and speed of signature-based approaches. Treephyler was evaluated on a real metagenome to assess its performance in comparison to previous approaches for taxonomic profiling.