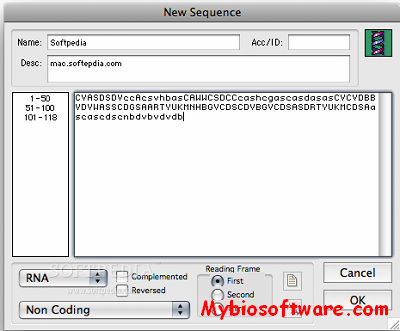
Se-Al is an application for creating multiple sequence alignments from nucleotide and amino acid sequences. At the moment it does not do any automatic alignments but is intended for the production of hand alignments and for preparing input for alignment programs such as CLUSTAL and phylogeny reconstruction programs such as PHYLIP and PAUP. It is particularly useful for manipulating protein coding DNA/RNA sequences.
goldMINER automates local and remote searches against pFAM and CDD (Conserved-Domain Database, which integrates pFAM, SMART and COG, etc.) databases searches to annotate sequences such as expressed sequence tags (ESTs). It is the first software package to use the power of CDD at NCBI to automate sequence annotation.
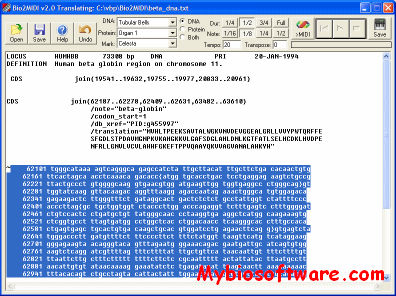
Bio2Midi converts the text of a DNA or protein sequence to a MIDI file, which you may immediately audition, or import into any MIDI sequencer for further compositional processing.
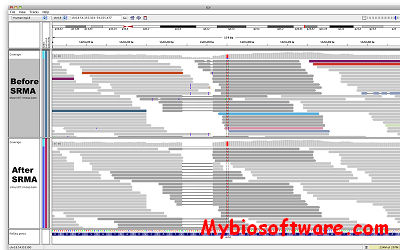
SRMA (Short Read Micro re-Aligner) is a software package that performs sequence re-alignment for next generation sequencing technology.It fixes artifacts introduced by aligning each read independently, significantly reducing the false positive detection, while cleaning insertions and deletions.
Evolver is a collection of programs designed to simulate the evolution of the nucleotide sequence of a whole genome. Evolver simulates the evolution of a representative genome of a species over periods long compared with its generation time.
::DEVELOPER
Robert Edgar , George Asimenos, Serafim Batzoglou and Arend Sidow.
CLOBB (Cluster on the basis of BLAST similarity) takes a set of DNA sequences and clusters them into groups which putatively derive from the same gene. In order to operate, the user must have BLASTALL in their path. The output is a blastable fasta file named <cluster_id>EST, where cluster_id is given by the user, which contails a list of sequences with identifiers <cluster_id>00001 to <cluster_id>99999.
::DEVELOPER
John Parkinson (john.parkinson@ed.ac.uk) and Mark Blaxter , Institute of Cell, Animal and Population Biology, University of Edinburgh
DIYA (Do-It-Yourself Annotator) is a modular and configurable open source pipeline framework, written in Perl, used for the rapid annotation of microbial genome sequences. The software is currently used to take nucleotide sequence contigs as input, either in the form of complete genomes or the result of shotgun sequencing, and produce an annotated sequence.