SARA is a very fast method for doing single side chain replacements in protein structures by using a coarse- grained method. It is over five times faster than the leading all-atom approach, and generates biologically realistic side-chain angles. The solutions found by SARA typically deviate less than 1 ? and 12 degrees from native structures or the best all-atom solution. Run-time for the algorithm is highly predictable and can easily be tuned by the user. These characteristics makes SARA an excellent choice for high-throughput applications like structural genomics, evolutionary simulations and structure-based phylogenetics.
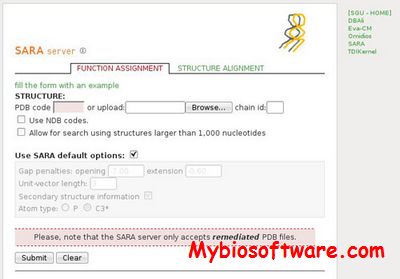
SARA is a program for pair-wise alignment of RNA structures as a web server for structure-based RNA function assignment. The SARA server relies on the SARA program, which aligns two RNA structures based on a unit-vector root-mean-square approach. The likely accuracy of the SARA alignments is assessed by three different P-values estimating the statistical significance of the sequence, secondary structure and tertiary structure identity scores, respectively.