BAIT (Bioinformatic Analysis of Inherited Templates) is a software to create strand inheritance plots in data derived from the Strand-Seq sequencing protocol. The software is designed to be flexible with a range of species, and basic template folders can called to read in species-specific data.
GRIL is a tool that can be used to identify the location of rearrangements and inversions in the backbone of a set of DNA sequences. GRIL works by identifying exactly matching regions present in all sequences under consideration and organizing them into groups of collinear regions. GRIL removes small regions of collinearity that appear unlikely to be true sequence rearrangements based on user-specified criteria such as the length of the collinear region and the percent sequence identity of the collinear region. The size of sequences that GRIL can be applied to is dependent on the amount of available memory.
Diogo Pratas, Raquel M. Silva, Armando J. Pinho, Paulo J. S. G. Ferreira.
An alignment-free method to find and visualise rearrangements between pairs of DNA sequences.
Scientific Reports, 2015 (Accepted).
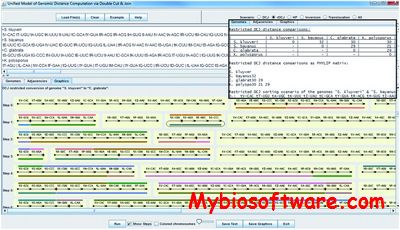
UniMoG (former DCJ) is a software tool unifying five genome rearrangement distance models: double cut and join (DCJ), restricted DCJ, Hannenhalli and Pevzner (HP), inversion only and translocation only. It allows computing all of these five distances between pairs of genomes represented as sequences of oriented common blocks.
DCJ (double-cut-and-join) computes the double-cut-and-join distance between two genomes and an optimal sorting scenario that transforms one genome into the other. It operates on the most general model of genomes with a mixed collection of linear and circular chromosomes. The sorting process includes all classical rearrangement operations such as inversions, translocations, transpositions, block interchanges, fusions and fissions.
The software DeCoLT computes adjacencies (or any type of relation, like regulation, interaction, functional relationships) between ancestral genes from gene phylogenies reconciled with a species phylogeny according to duplications, losses and lateral gene transfer. It takes as input (1) a species tree (2) a set of extant genes (3) a set of exant adjacencies (relations) between extant genes and (4) reconciled gene trees which leaves are the extant genes.
DRIMM algorithm extends the GRIMM-Synteny (Genome Rearrangements in Men and Mice) algorithm towards the problem of identifying the synteny blocks in highly duplicated genomes.