SPECTRE provides several new implementations of pre-published algorithms to construct phylogenetic trees and networks (more precisely split networks), along with an interactive graphical interface for visualizing planar split networks.
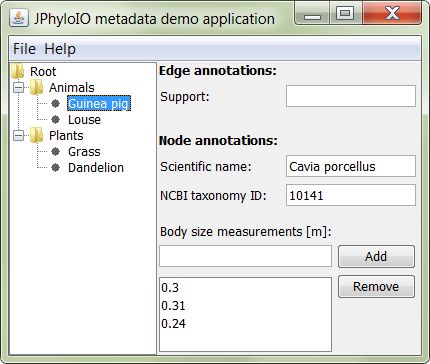
JPhyloIO is an open source Java library for reading and writing phylogenetic file formats. The main aim is to provide access to various formats using a single interface, while being independent of the concrete application data model, to achieve maximal flexibility.
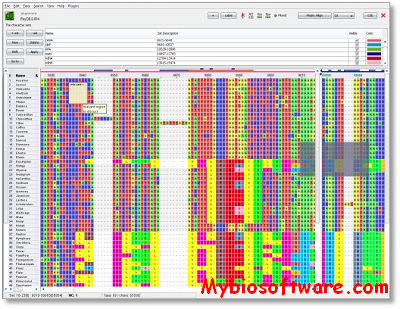
PhyDE (Phylogenetic Data Editor) is a system-independent editor for DNA and amino acid sequence alignments, designed to assist anybody interested in phylogenetic or other comparative analyses of sequence data.
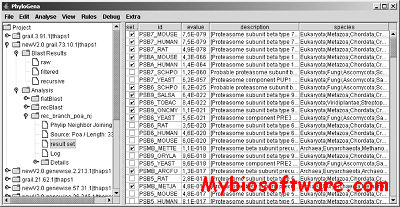
PhyloGena is a software tool to facilitate phylogenetic annotation of unknown sequences. It has a user-friendly graphical interface and you will intuitively learn how to use it. You can import 1, 10, 100, 1000? (see below) DNA or protein sequences and the program will search for similar sequences, construct a multiple alignment and subsequently a phylogenetic tree for all of them and show them to you. You can easily manipulate the data sets, add or remove sequences, change parameters etc. This is of great help in identifying the function and phylogenetic affiliation of ORFs and makes annotation of genes or ESTs easier and less error-prone.
DILTAG is an algorithm allowing to infer a set of optimal evolutionary histories for a gene cluster in a single species, according to a general cost model involving variable length duplications (in tandem or inverted), deletions, and inversions.
MaxAlike algorithm aims at reconstructing a nucleotide sequence in a target species, based on a multiple sequence alignment and a phylogenetic tree of homologous sequences from other species. For each alignment position the probabilities of occurrence for each nucleotide are computed, considering the phylogenetic position of the target species in the tree.
PhyloSort is a Java tool to sort phylogenetic trees by searching for user-specified subtrees that contain a monophyletic group of interest defined by operational taxonomic units (OTUs).
ExAlt is a software program designed to predict alternatively spliced overlapping exons in genomic sequence. The program works in several ways depending on the available input. ExAlt can use information about existing gene structure as well as sequence conservation to improve the precision of its predictions. ExAlt can also make predictions when only a single genomic sequence is available.