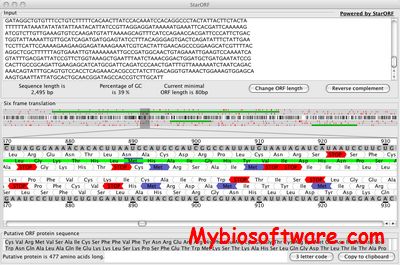
StarORF facilitates the identification of the protein(s) encoded within a DNA sequence. Using StarORF, the DNA sequence is first transcribed into RNA and then translated into all the potential ORFs (Open Reading Frame) encoded within each of the six translation frames (3 in the forward direction and 3 in the reverse direction).
TAMO (Tools for Analysis of MOtifs) is developed around a unified motif representation of a position-specific scoring matrix (PSSM). Motif objects may be assembled from IUPAC-ambiguity codes, multiple sequence alignments, averages of other motifs, and matrices of frequencies or log-likelihood values. Motifs can printed, concatenated, indexed and sliced like text strings, or rendered as sequence logos. They can also be randomized, reverse-complemented, and recomputed using different assumptions about background base frequencies. Motifs can also store and report information about their origin, information content, and score. Finally, motifs can scan DNA sequences for instances of matching sites.
ThioFinder is a web-based tool to rapidly identify thiopeptide biosynthetic gene cluster from DNA sequence using a profile Hidden Markov Model approach.
IMMpractical implements various Markov chain model-based methods for analysis of DNA sequences.Markov chain models are commonly used for content-based appraisal of coding potential in genomic DNA.
RHOM (Research of HOMogeneous regions in DNA sequences) is software designed for the use of hidden Markov chain models for the segmentation of DNA sequences in homogeneous regions. R’HOM makes it possible to estimate a more realistic model of DNA sequence composition than a homogeneous Markov chain model and then to segment the sequence under this model. It has been used in particular to look for horizontal transfers in B. subtilis and to estimate models designed to calculate the significance of word counting. R’HOM has been developed in cooperation with the Laboratoire Statistique et Génome in Evry.
SPADS is a population genetics software computing several summary statistics from populations or groups of populations, and implementing two clustering algorithms to study the genetic structure of populations. SPADS has been specifically developed for the analysis of DNA sequences. The first aim of SPADS is to compute, on real datasets, the summary statistics computed by PhyloGeoSim on each simulated datasets.
seqRFLP includes functions for handling DNA sequences, especially simulated RFLP and TRFLP pattern based on selected restriction enzyme and DNA sequences.
Sigma (Simple greedy multiple alignment) is an alignment program with a new algorithm and scoring scheme designed specifically for non-coding DNA sequence.This problem is now growing in importance with the increasing number of fully-sequenced species. In particular, studies of gene regulation seek to take advantage of comparative genomics, and recent algorithms (such as PhyloGibbs) for finding regulatory sites in phylogenetically-related intergenic sequence require alignment as a preprocessing step.
TESS (Transcription Element Search System) reads (selected) PWMs (Partial Weight Matrices) from a file and predicts binding sites on DNA sequences read from another file.