rapmad is an R-package for the Robust Analysis of Peptide MicroArray Data. It is an automated, multi-step approach that combines several computational and statistical procedures to improve the quality of peptide microarray data and thus enable a more reliable analysis.
SHORE is a mapping and analysis pipeline for short DNA sequences produced on Illumina Genome Analyzer and Hiseq 2000, Life Technology SOLiD, 454 Genome Sequencer FLX and PacBio RS platforms. It is designed for projects whose analysis strategy involves mapping of reads to a reference sequence. This reference sequence does not necessarily have to be from the same species, since weighted and gapped alignments allow for accuracy even in diverged regions. SHORE provides various prediction algorithms for genomic polymorphisms, i.e. SNPs, structural variants (indels, CNVs, unsequenced regions), SNPs and SV prediction in heterozygous or pooled samples, as well as peak detection for ChIP-Seq analysis and quantitative analysis of mRNA-Seq and sRNA-Seq.
SNPTools is a suite of tools that enables integrative SNP analysis in next generation sequencing data with large cohorts. It not only calls SNP in a population with high sensitivity and accuracy, but also employs a novel imputation engine to achieve highly accurate genotype calls in an efficient way.
popoolation 1.2.2 / PoPoolation2 1.201 / PoPoolation TE 1.02
:: DESCRIPTION
PoPoolation is a collection of tools to facilitate population genetic studies of next generation sequencing data from pooled individuals
PoPoolation2 allows to compare allele frequencies for SNPs between two or more populations and to identify significant differences.
PoPoolation TE is a quick and simple pipeline for the analysis of transposable element insertions in (natural) populations using next generation sequencing.
MUFFINN (MUtations For Functional Impact on Network Neighbors) is a method for prioritizing cancer genes accounting for not only for mutations of individual genes but also those of neighbors in functional networks
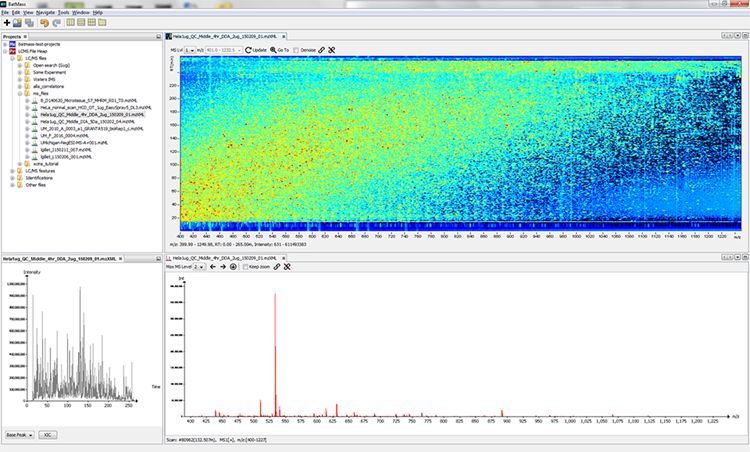
BatMass is a mass-spectrometry data visualization tool, with the main focus on being fast and interactive while providing comprehensive visualizations without any parameter tweaking.
HapSeq2 is a program for genotyping calling and haplotype phasing from next generation sequencing data using haplotype information from jumping reads. Previously, we developed a Hidden Markov Model (HMM) based method for genotype calling and haplotype phasing from next generation data that can take into account jumping reads information across two adjacent potential polymorphic sites.
ChromatoGate (CG) has been created to accelerate the process of detecting possible errors in DNA sequences that have been introduced by Sanger sequencers. To detect possible errors in the sequences, CG starts from the multiple-sequence alignment instead of inspecting every sequence separately.
HiTEC (High Throughput Error Correction) , an algorithm that provides a highly accurate, robust and fully automated method to correct reads produced by high-throughput sequencing methods. The approach provides significantly higher accuracy than previous methods. It is time and space efficient and works very well for all read lengths, genome sizes and coverage levels.
NGC is a compressor for aligned HTS sequencing data that enables the complete lossless and lossy compression of mapped alignment data stored in SAM/BAM files.