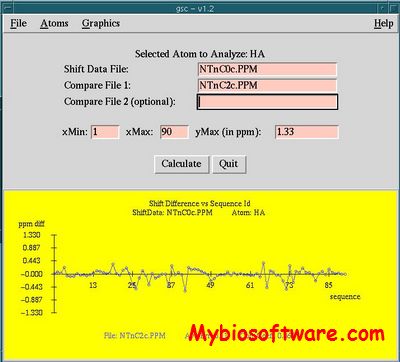
RNACluster is an integrated computational software which implements 6 common structure distances to measure the (dis)similarity of RNA secondary structures including base pair distance, mountain distance, morphological distance, tree edit distance, string edit distance and our in-house structure matrix distance, and one effective cluster approach for the ensemble clustering using a minimum spanning tree (MST) based algorithm. RNACluster can be used to study the characteristics of RNA secondary structures, RNA structure conformational switches, RNA conformational energy landscapes and RNA secondary structure prediction based on the clustering of structure ensemble.
MUTPROF is a Monte-Carlo-Markov-Chain implementation of the hypergeometric test for sparse 3-dimensional contingency tables (in the spirit of Fisher’s exact test). It allows the comparison of up to 200 positions within the same gene, but between different tissues and/or species.
MUTCOMP serves to compare two nucleotide substitution matrices for one or more, possibly different genes, tissues or species, taking the expected mutation frequencies into account.
STRuster is a method for clustering alternative structural models corresponding to different structure determination experiments. Alternative structural models, determined by X-ray crystallography or NMR spectroscopy, are frequently available for a given protein. These models can present significant structural dissimilarity. The structures are classified according to backbone structure similarity.
CLICK is capable of superimposing the 3D structures of biomolecules, the Cartesian coordinates of whose constituent atoms are presented in the PDB format. In addition to coordinates, the web server can make use of similarity of other structural features such as secondary structure, solvent accessible surface area, and residue depth to guide the alignment. CLICK first looks for cliques of points (3–7 residues) that are structurally similar in the pair of structures to be aligned. Using these local similarities, a one-to-one equivalence is charted between the residues of the two structures. A least square fit then superimposes the two structures. Our method is especially powerful in establishing protein relationships by detecting similarities in structural subdomains, domains and topological variants.
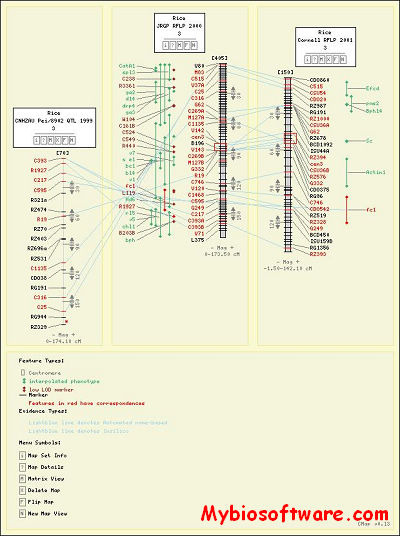
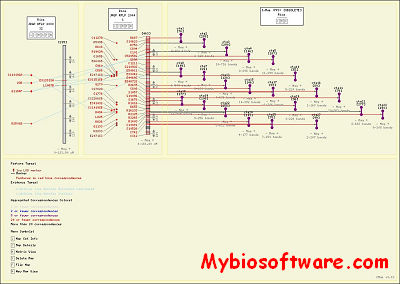
CMap (omparative Map Viewer) is a web-based tool that allows users to view comparisons of genetic and physical maps. The package also includes tools for curating map data. A user can compare an unlimited number of maps, view pair-wise comparisons of known correspondences, and search for maps or for features by name, species, type and accession. CMap is freely available, can run on a variety of database engines and uses only free and open software components.