FNV (Flashed-based Network Viewer) is for the visualization of small to moderately sized biological networks and pathways. FVN can also be used to embed pathways inside PDF files for the communication of pathways in soft publication materials
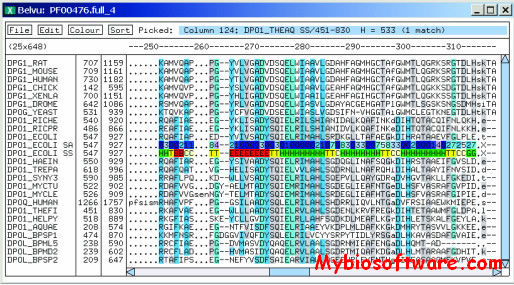
Belvu is an X-windows viewer for multiple sequence alignments. One of the main advantages of Belvu is that it has an extensive set of modes to color the residues. There are several ways to color them by conservation and by residue type (user-configurable). Other useful features are fetching of the Swissprot (or PIR) entries by double clicking and easy tracking of the position in the alignment.
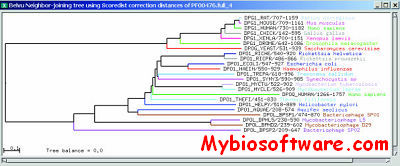
In addition, Belvu is a phylogenetic tool. It can be used to generate distance matrices between sequences under a selection of distance metrics. These can be saved and used subsequently in other applications. Belvu also implements certain distance-based tree reconstruction algorithms – including import of externally generated distance matrices – and bootstrap phylogenetic reconstruction. These functions are available both in the GUI (meaning Belvu may also be used as a tree viewer) or as command-line options, making the program a potential component in phylogenetic software pipelines.
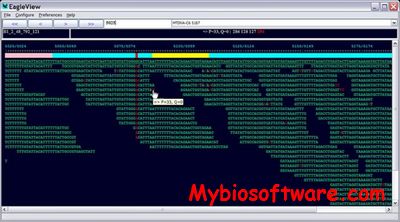
EagleView is an information-rich genome assembler viewer with data integration capability. EagleView can display a dozen different types of information including base qualities, machine specific trace signals, and genome feature annotations. It provides an easy way for inspecting visually the quality of a genome assembly and validating polymorphism candidate sites (e.g., SNPs) reported by polymorphism discovery tools. It can also facilitate data interpretation and hypothesis generation.
G Yu, DK Smith, H Zhu, Y Guan, TTY Lam*.
ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data.
Methods in Ecology and Evolution. 2017, 8(1):28-36
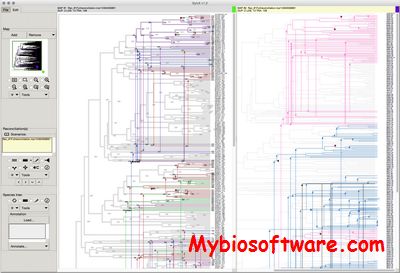
SylvX is a reconciliation viewer which implements classical phylogenetic graphic operators (swapping, highlighting, etc.) and new methods to ease interpretation and comparison of reconciliations (multiple maps, moving, shrinking sub-reconciliations).
JSAV is a sequence alignment viewer written purely in JavaScript. It is extremely easy to include it in your code, but has many configuration options allowing you to control the functionality available to the user of your web site.
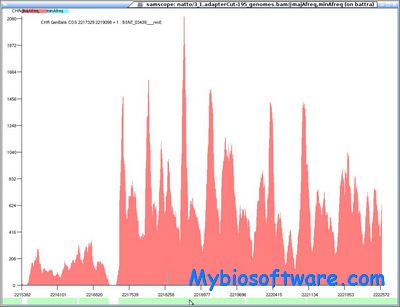
Samscope is a lightweight SAM/BAM file viewer that makes visually exploring next generation sequencing data intuitive and maybe even fun!Samscope uses multiple layers to simultaneously (or sequentially) view SAM/BAM related features like coverage or allele frequency, or ChIP-SEQ features like polarity. The paging-friendly binary file layout makes it feasible to browse data sets far larger than the system’s available RAM.